Toward a Silicate-Based Molecular Nanotechnology
I. Background and Review
by
Stephen L. Gillett
Dept. of Geological Sciences
Mackay School of Mines, University of Nevada
Reno, Nevada 89557 USA
e-mail: [email protected]
Based in part on a presentation given at the
Fifth
Foresight Conference on Molecular Nanotechnology. Palo Alto, California, 8 Nov 1997.
This version was submitted 15 Dec 1998.
Abstract
Although theoretical studies of molecular nanotechnology (MNT) have focused on tetrahedral carbon ("diamondoid") structures, analogous silicon structures are an unpromising basis for MNT, despite the importance of Si in semiconductor technology. The resemblances of Si and C chemistry are largely formal; in particular, the Si-Si bond is not nearly so stable as is C-C, and Si shows minimal tendency to form the double and delocalized bonds so typical of C. Polymeric silicates, however, compounds characterized by disiloxy (Si-O-Si) linkages, show promise for MNT, because of:
- The strong and directional character of the Si-O bond, due to its high stability and partly covalent character.
- Their ease of polymerizing into 3D structures ("tectosilicates"). Silicates enter tetrahedral coordination at STP, even out of aqueous solution, in stark contrast to sp3 carbon.
- Their thermal stability: certain tectosilicates, such as quartz and feldspars, will even crystallize directly from silicate melts at hundreds of degrees C.
- Their stability to oxidizing conditions; whatever its other virtues, C burns.
- Their sheer abundance. O and Si make up, respectively, 60.4 and 20.5 atomic percent of the Earth's crust. Nearly all common minerals are silicates.
Silicate-based nanotechnological structures are likely to be particularly valuable where an unreactive void-bearing framework is desirable, as in selective catalysis, ion-exchange and sorption, solid electrolytes, and as a supporting host for such applications as arrays of semiconductor clusters for novel electronic and optical devices. The theoretical strength of silica also compares favorably with other ultrastrong materials.
Silicates at ordinary pressures are based on an SiO44- tetrahedron; these tetrahedra can share one through all four vertices to build up a vast array of structures, including chains, sheets, and infinite 3D networks. They thus in essence form giant-molecule anions in which charge balance is preserved by cations that fit between the anionic structures. The diversity of structures is further broadened by substitution of other atoms for Si. In natural systems, Al substitution is ubiquitous; one class of alumino-tectosilicates, the feldspars, are the most common compounds in Earth's crust. Other alumino-tectosilicates, the zeolites, have great technological importance as "molecular sieves" and catalysts, due to their molecular-sized internal voids. Furthermore, siloxanes ("silicones"), "hybrids" of silicates and organic compounds that have H or organic side-groups attached to Si show how silicate-based structures could be integrated with C-based structures. Water-soluble silicates, which are promising raw materials for synthesis, consist of mixtures of oligomers, polymers, and colloids, and colloidal sols can be rendered metastable with high SiO2 content at relatively low pH.
Though silicate raw materials are literally everywhere, conventional mining waste seems especially attractive, as it is both comminuted and otherwise presents serious disposal and storage problems. Considerably less energy-intensitive extraction technologies for silicate raw materials also seem possible based on recent chimie douce approaches. Silicate-based MNT also has major potential applications in space development, as many extraterrestrial bodies (e.g., the Moon) are dominated by silicates but have scant carbon.
Silicate chemistry has been historically fragmented and was worked out only in the second quarter of the 20th century, which may account for its lack of familiarity to nonspecialists; nonetheless, it seems to offer broad scope for nanotechnological endeavors, and may well prove easier to implement in the near term.
1. Introduction.
"... the silicates, more than any other group of chemical compounds with the exception of the organic carbon compounds, exhibit an enormous chemical and structural variability."
-- F. Liebau, 1985 (p. 266)
Theoretical studies of molecular nanotechnology (MNT) have focused on tetrahedral carbon ("diamondoid") frameworks (Drexler, 1992, p. 13; Merkle, 1995). This focus is motivated by the propensity of carbon to link to itself to form arbitrarily complex molecular structures, as is shown by the enormous range of both natural and synthetic organic chemistry. The existence of C-based molecular machinery in biosystems and the great strength/mass ratio theoretically achievable with diamondoid frameworks, due to their strong and directional covalent bonding, furnish further motivations. The enormous importance of organic chemistry has also made molecular modeling reasonably routine. Finally, of course, carbon is a fairly common and readily available element at the Earth's surface.
It seems natural to wonder, though, whether silicon, carbon's second row homolog in the periodic table, could form a reasonable basis for MNT, especially given the importance of Si in semiconductor technology. However, Si itself is unpromising: the Si-Si bond is not nearly so stable as is C-C, and Si shows minimal tendency to form the double and delocalized bonds so typical of C. Indeed, as discussed below, the resemblances between Si and C chemistry are largely superficial.
Nonetheless, an Si-based MNT seems not merely plausible but promising. However, it would not be based on Si itself but on silicates, compounds characterized by Si-O linkages. Silicates, substituted silicates, and certain similar compounds can form relatively inert framework structures consisting basically of tetrahedra of oxygen anions surrounding a small central cation (Si, Al, or other; a "T atom"). These tetrahedra then share vertices to form tetrahedral frameworks (T-frameworks), linked by bridging oxygens, whose strength and flexibility seem promising as a basis for MNT structures.
Despite being somewhat weaker than diamondoid structures, such structures have many potential advantages for nanotechnological devices:
- Silicates are stable in an oxygen atmosphere, as obviously they cannot oxidize.
- Silicate frameworks can be much more heat-tolerant than organic frameworks. The frameworks of certain natural and artificial zeolites, the prototypes of such compounds, can remain intact up to hundreds of degrees C. Indeed the name "zeolite" comes from the Greek for "boiling stone", in reference to the fact that included water in intracrystalline voids can be boiled out.
- Many silicates are largely inert to common reagents, as is shown by the ubiquitous use of silicate glass vessels in conventional wet chemistry. Indeed, some SiO2-rich structures are chemically attacked only under extreme conditions, e.g., by HF or at extreme pH (McDaniel & Maher, 1976).
- Silicates have a degree of structural and compositional flexibility that compares favorably with that of diamond-based structures, particularly when Si is substituted with other tetrahedrally coordinated atoms (Al, B, etc.).
- 3D polymerization of silicate structures can occur spontaneously at room temperature, even out of aqueous solution. This contrasts with diamondoid structures, which require either extreme conditions or sophisticated nanomechanical assemblers that do not yet exist. Hence synthesis may be much more straightforward.
- Substituted silicate frameworks may be readily fabricated to have a net electrical charge. Indeed, many natural minerals are aluminosilicates, in which substitution of Al for Si yields a polymeric aluminosilicate anion "backbone." Such charged frameworks have many potential applications in ion exchange, transport, and catalysis, and seem more difficult to achieve with diamondoid structures.
- Silicates and substituted silicates can easily form frameworks that support voids of molecular and supramolecular size, often called "molecular sieves." Although presumably a diamondoid framework could support large voids easily enough, and indeed such voids may be an unwanted by-product of certain synthesis pathways (Krummenacker, 1994), a silicate framework should nonetheless offer advantages in many applications, the resistance of the framework to oxidation being one obvious example. Tailoring of the voids' chemical environment through T-atom substitution, as is currently the goal of much zeolite research to improve catalysis, is another.
- Finally, silicates are extremely abundant: silicates and aluminosilicates make up nearly all the Earth's crust (and any rocky planet's crust, for that matter; "rocks" are silicates), as O (60.4 atom %), Si (20.5%), and Al (6.3%) are the most abundant elements in the crust (e.g., Mason, 1966). Carbon, by contrast, is substantially less abundant, and on some other rocky bodies in the Solar System (e.g., the Moon) it is essentially absent. Hence, resources are literally everywhere.
Silicate chemistry, however, is not nearly so familiar to the nonspecialist as is organic chemistry. Not only does the superficial similarity of C and Si tend to lead the unwary into consideration of molecular structures containing Si-Si bonds, but it leads to overlooking the significance of the disiloxy (-Si-O-Si-) bond. In addition, silicate chemistry also lacks the clear historical focus of organic chemistry, because it was worked out only in the 20th century and is furthermore scattered across a number of disciplines, including mineralogy, ceramic chemistry, inorganic chemistry, and catalysis.
This paper will gather together some of these scattered threads and highlight their likely relevance for MNT for the benefit of a nonspecialist audience. References cannot be comprehensive, but are intended to provide an entry point to the literature. A following paper reviews silicate molecular modeling, gives a brief overview of the current syntheses of silicate structures, and sketches some possible approaches to silicate molecular assembly.
2. Some Potential Applications
Several potential near-term nanotechnological applications for silicate or substituted-silicate frameworks are described briefly below.
2.1 Materials.
As ordinary rocks are largely composed of silicate minerals, silicate-based materials are already of great technological importance; nonetheless, their potential usefulness extends well beyond their value as such bulk industrial commodities such as sand, gravel, and concrete aggregate.
Glass, a disordered 3D silicate polymer, is of course already ubiquitous, but its vulnerability to shattering hardly needs emphasis. Indeed, the brittleness of traditional silicate building materials, such as building stone, also has limited their use in modern structures to largely decorative uses, though their non-flammability has also been important. As with other conventional brittle materials, the great potential strength of the Si-O bond is unrealized with present-day fabrication techniques, owing to their lack of molecular-scale control. Instead, silicate materials are exceedingly vulnerable to catastrophic Griffith failure due to their high density of microflaws. A motivation for MNT is the hope of achieving bulk materials with strengths approaching the limits set by chemical bonds; silicates are particularly desirable candidates because of their potential strength, their abundance, and their non-flammability.
Crystalline silicates would also benefit from molecular-level assembly, for such applications as optics and electronics. Quartz, for example, is the most stable crystalline form of silica (SiO2) at STP and belongs to a rare non-centrosymmetric crystal class (32). This accounts for its value in piezoelectric applications.
2.2 Molecular frameworks and sieves.
Silicate MNT is particularly likely to be useful where an unreactive void-bearing structure is desirable. The zeolites and related "molecular sieves" are perhaps the most outstanding examples. These have a sufficiently open framework to support molecular-sized voids and passageways. Some particularly important applications for such structures are listed below:
2.2.1 Gas sieves/ion exchange.
Both of these are classic zeolite applications. Steric hindrance, in which the channels in a T-framework structure allow some molecules to pass while blocking others, can be used to separate molecules. For example, O2 can be removed from air at ambient temperature by Na-zeolite A to yield a stream enriched in nitrogen (Szostak, 1989, p. 17-18), and such separations find use in state-of-the-art air purifiers, such as those used in military applications. As N2 and O2 molecules differ little in size, this indicates the degree of selectivity possible in a T-framework and justifies the name "molecular sieve." Other molecular separations are also important in industry (e.g., Flanigen, 1991).
In fact, the separation of different kinds of atoms and molecules from one another is a profound technological problem--consider pollution control and resource extraction--and it may well be one of the early drivers toward MNT (Gillett, 1996a). T-framework structures promise many applications along this line, and macroscopic, nano-tailored zeolitic constructs would be vastly more useful than those presently available. Removal of O2 from air, for example, would be much more practical if truly macroscopic "sieves" were available. The present sieves consist of microcrystalline powders: they must be added like reagents to absorb passing molecules, and so must be reactivated (and replenished) frequently. A macroscopic molecular sieve that could separate gas mixtures by "straining" different molecules out would be considerably more efficient than the thermal fractionation presently used for large-scale separation of gases. Indeed, a "membrane" consisting of a zeolitic structure extended indefinitely in two dimensions, is an obvious possibility; current "ceramic membranes" are seriously limited in their applications because (like other membranes) they are impossible to fabricate atomistically.
Zeolites were also the first ion-exchangers, and still find application here, especially in detergents (Townsend, 1992). As described below, zeolites are characterized by an anionic aluminosilicate framework that includes charge-compensating cations in the intracrystalline voids. These cations can exchange with others in solution, and the selectivity of the exchange can be high both because of steric selection, and because of subtle differences in the electrostatic binding of different cations to the framework. Again, the potential of such materials for the selective extraction of particular dissolved species is obvious.
2.2.2 Solid electrolytes.
Solid electrolytes conduct ions while remaining in the solid state (e.g., Rickert, 1982). They are useful for fuel cells (e.g., Murugesamoorthi et al., 1993), because they can allow the intimate, controlled reaction of fuel and oxidizer as charged species without the need for a liquid phase. A related use might be an "ion pump", for extracting ions from liquid solution.
Solid electrolytes work by having many ionic site vacancies. Under the influence of an electric field an adjacent ion can hop into the vacancy, leaving a new vacancy behind. Thus in effect the vacancy moves, analogous to the motion of a "hole" in a semiconductor. Obviously (and unfortunately), however, this hopping works best at high temperature, where the thermal motion of the ions makes them easier to move. For example, one of the most practical solid electrolytes is presently doped ZrO2, but it is only effective > 600°C (e.g., Rickert, 1982). (The crystal fundamentally consists of close-packed O2- ions, which include the Zr4+ cations in the interstices. Substitution of (e.g.) ~10% Y3+, instead of Zr4+, however, leaves some O2- sites vacant to preserve charge balance.) Such high-T operation obviously severely limits potential applications, especially exotic ones such as ion pumps that must work near ambient temperatures.
Zeolites have been investigated as alternative solid electrolytes (e.g., Kelemen et al., 1989), but currently accessible structures are impractical, as they have conductivities several orders of magnitude lower than alternative ionic conductors. Some rare-earth-bearing sheet-silicate structures show promising ionic conductivities (Haile et al., 1993), however, as do certain phosphates and phosphosilicates with open-channel framework structures such as Nasicon (Corbridge, 1995, p. 292-294, and refs. therein). Shannon et al. (1978) also reported promising Na+ ionic conductivity in a rare-earth silicate containing Si12O3624- anionic rings. Hence, it seems likely that silicates could be tailored so that ions would be much more mobile, and thus the solid electrolyte work at far lower temperatures, than the currently known structures.
2.2.3 Catalysts/fixed acids.
This is probably the most important present-day application of zeolites (e.g., Newsam, 1986) and continues to motivate a great deal of research, especially in the petrochemical and refining industries. Active catalytic sites in the zeolite voids can only operate on molecules that can fit through the openings into the zeolite structure. This yields a catalytic specificity that commonly invites comparison with biological enzymes (Herron, 1989). Indeed, zeolites can be considered as analogous to proteins in providing a constrained environment for the controlled reaction of active molecules (e.g., Turro & Garcia-Garibay, 1991).
Moreover, crystalline solids such as zeolites are potentially more practical for catalytic applications because, rather than being in solution with the reactants as are most enzymes, they remain separate and immobile; thus they need not be separated from the solution after reaction is complete (e.g., Thomas, 1992). Indeed, some authors call the zeolitic voids "nanoreactors" (e.g., Pool, 1994): uniform, nanometer-scale vessels in which highly specific reactions can occur. This in turn suggests such nanovessels may ultimately have a role in molecular assemblers. (Note, after all, that silicates make up most macroscopic labware!)
Most such catalysts are "fixed" acids, in which the charge-balancing cation is an H+ hydrogen-bonded to a bridging oxygen in the T-framework (e.g., Szostak, 1989, p. 30-39). They have an extremely wide array of applications, especially in hydrocarbon refining (Jacobs & Martens, 1992); the cracking of specific alkanes (which recently has been modeled on an ab initio basis; Collins & O'Malley, 1995), and gasoline synthesis from methanol (e.g., Thomas, 1992) are merely two examples.
Although fixed acids dominate current catalytic applications, zeolite catalysts containing included metal cations rather than H+ have been used (e.g., Flanigen, 1991) and remain promising for specialized applications (e.g., Newsam, 1986; Feng et al., 1997ab).
Related to such applications is electrode modification (Rolinson, 1990; Bard & Mallouk, 1992). In this case, the size and shape of the voids in the zeolite limit access to the electrode surface to only certain molecules. This is useful in building exquisitely sensitive sensing systems for particular solutes, and may also prove useful in electrochemical syntheses such as electrocatalysis.
2.2.4 Inert supporting structures.
Zeolites have been used experimentally as supporting substrates for molecular chemical systems (e.g., Ozin, 1992; Stucky, 1992), including such things as artificial photosynthesis (e.g., Persaud et al., 1987; Krueger et al., 1988; Li et al., 1988; Ramamurthy, 1991; Anpo et al., 1997). They have also been used in 3D "ship in a bottle" (Bard & Mallouk, 1992; Cano et al., 1996) constructions, in which "guests" (e.g., atomic clusters, organic molecules) are dispersed in the framework voids (e.g., Jelinek et al., 1991). Included metal or semiconductor clusters (e.g., CdS; Herron et al., 1989), for example, promise major new applications in electronics and nonlinear optics (Ozin et al., 1989; Ozin & Öskar, 1992). Because the spacing between the clusters can be varied, depending on the size and spacing of the zeolite voids, the electronic properties due to the clusters' interactions can be changed vastly. Zeolites are also being investigated as supports for molecular conductors ("nanowires"), which would consist of conducting organic polymers threaded through the zeolite channels (Bein & Enzel, 1993). Such constructions could lead to a vast new array of miniature electronic devices.
In fact, such research suggests zeolites may have a role in "bootstrapping" more sophisticated molecular assemblers. Even though zeolite crystals made by conventional synthesis techniques are small in everyday terms (~1 µm), they are still roughly 104 atoms across. Hence such relatively large, atomically precise, void-bearing structures may be useful as substrates for further nanotechnological assembly.
The inertness of the Si-O-Si framework is especially a consideration in these applications, as there is minimal interaction of the framework with the guests; they instead are free to interact with each other. This may be more difficult to arrange for a diamondoid substrate, at least for applications involving organic molecules. Indeed, in a T-framework the attached molecules need not be "bound" at all, at least with conventional covalent bonds; they can be merely trapped by steric effects (as with guests or threaded polymers), possibly in conjunction with van der Waals forces or hydrogen bonds, as with an H to a bridging oxygen. Electrostatic bonds to the sites in the T framework that are normally occupied by the charge-compensating cations would also be relatively easy to arrange.
2.2.5 Combinations.
Of course, with sufficiently sophisticated molecular assembly, "combination" applications can be envisioned. For example, consider a "solute extractor and sorter": nanowires threaded through some of the channels in a T-framework direct "ion pumps" through other channels, perhaps by changing the local electrostatic environment in the channels in a controlled way so that ions are moved along them systematically. In this way it might be possible to extract specific ionic species from a solution. In addition, uncharged solutes could possibly be extracted. They could certainly be preferentially adsorbed into correctly shaped voids, as in the current applications involving gas sieving. Possibly such absorbed species could then be moved electrically as well, e.g., by induced polarization. (Detailed modeling is obviously required here.) The utility of such a nanodevice in (say) pollution control and remediation is obvious.
Finally, note that all such applications are nanostructured, not "nanomechanical"; i.e., they have no moving molecular parts except individual charged species. Elsewhere (Gillett, 1996b) I have suggested that assembly of such nanostructured materials is likely to be the "breakthrough" application of nanotechnology. Not only does fabrication seems much easier, as self-assembly approaches may be possible, but the absence of molecular moving parts suggests they will have greater reliability.
3. Silicate Chemistry, I. Silicon vs. Carbon
Despite their adjacent position in Group IV of the Periodic Table, the chemistry of Si and C contrasts markedly, and fundamentally, silicon chemistry cannot be understood by referring to carbon chemistry! The fundamental reason, as for the differences between other first- and second-row elements, is the greater size of the second-row atom.
First, the multiple and delocalized bonds so typical of C chemistry are essentially absent. The Si atom is too large for the 3p orbitals to have much overlap, and hence p bonds are unstable. Although Si=Si double bonds have been synthesized, they remain largely a laboratory tour de force (e.g., Raabe & Michl, 1989). Not only are such compounds highly reactive, they must be kinetically stabilized by such subterfuges as bulky side chains to inhibit autodecomposition; in addition, despite many attempts, no Si=O double bonds or delocalized bonds have been synthesized except fleetingly, in stark contrast to C=O. Thus the multiple and delocalized ("aromatic") bonds so characteristic of carbon chemistry are essentially absent. However, the Si-O single bond is very stable, more so than C-O (Table 1).
Table 1. Bond Energies, Si and C
| |
|
Si |
|
C |
| |
|
kJ/mol |
aJ |
|
kJ/mol |
aJ |
| -Si |
|
322 |
0.535 |
|
372 |
0.618 |
| -O |
|
535 |
0.889 |
|
346 |
0.575 |
| =O |
|
-- |
|
808 |
1.343 |
| -C |
|
372 |
0.618 |
|
335 |
0.556 |
| -H |
|
376 |
0.625 |
|
404 |
0.671 |
| Table 1. Important Si and C bond energies. Si-X values from Barton & Boudjouk (1990); C-X values from table in Drexler (1992, p. 52). |
Second, Si prefers higher coordination numbers than C. Tetrahedral coordination with oxygen is usual at STP, and a small amount of 5-coordinate Si even occurs in glasses (Stebbins, 1991). Penta- and hexa-coordinate Si are common with certain ligands, as with fluoride and certain organics (e.g., Laine et al., 1991; Chuit et al., 1993). Hexacoordinate Si is also ubiquitous at high pressure (Finger & Hazen, 1991), and in any case hypercoordinate Si complexes seem to be extremely important as reaction intermediates (e.g., Liebau, 1984; Holmes, 1990; Kubicki et al., 1993). Finally, Si also has 3d orbitals available, and although their participation in Si bonding is evidently minor, it is non-negligible (e.g. Newton, 1981; Cruickshank, 1985; De Almeida & O'Malley, 1991; Gibbs et al., 1994).
These contrasts between the chemistry of C and that of Si are underscored by their oxides: although SiO2 and CO2 have the same stoichiometry, their structures could hardly differ more. CO2 is a discrete molecule in which the oxygens are bound with double bonds; p-bonding is possible between C and O because their 2p orbitals overlap. By contrast, SiO2 is an infinite 3D framework, occurring in a number of polymorphs, in which the oxygens bridge between adjacent Si atoms by partly covalent, single bonds. Though a hypothetical silica-type form of CO2 has been studied theoretically (Gibbs et al., 1988), it has never been synthesized and may well be impossible.
Another contrast is shown by the limited tendency of Si to form Si-Si bonds, in contrast to the ubiquity of C-C bonds. Although formally the energy is similar to C-C (Table 1), Si-Si is vulnerable to nucleophilic attack, probably because of its larger size and thus the availability of a hypercoordinate reaction intermediate. Similarly, although the Si-H bond is not much weaker than C-H (Table 1), it is considerably more polar and hence generally more reactive. The silicon analog of methane (SiH4, silane), for example, is much more reactive than methane, especially with oxygen. Finally, and curiously, the strength of the Si-C bond is comparable to that of C-C (Table 1) and is relatively unreactive; as will be seen, this fact has major implications for possible ways by which silicate and diamondoid structures could be integrated.
3.1 The Si-O bond.
The Si-O bond has ~50% covalent character (e.g., Liebau, 1985, pp. 46-48). The anomalously short (161 pm) Si-O distance in the SiO4 tetrahedron had originally been interpreted as reflecting a degree of 3d-hybridization (Cruickshank, 1961), although "anomeric" resonance (partial occupancy of an antibonding orbital by a lone pair on an adjacent electronegative atom) has been suggested instead (Reed et al., 1988; De Almeida & O'Malley, 1992ab). Recent ab initio modeling of H4SiO4 (De Almeida & O'Malley, 1991) and H6Si2O7 (O'Keeffe & McMillan, 1986) indicate that only a small amount of 2p-3d hybridization is present. Cruickshank (1985) concluded that although his original model overestimated the d-character, the participation of d-orbitals cannot be neglected in detailed models; as seems intuitively reasonable, second-row elements are intermediate between those of the first row, in which d participation is unimportant, and transition metals in which it is very important. Indeed, in detailed ab initio modeling of silicates, d orbitals prove necessary (Gibbs et al., 1994).
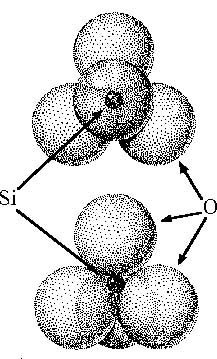 |
| Figure 1. The silicate tetrahedron, SiO4. In polymeric structures, adjacent tetrahedra share an oxygen at a vertex to form a disiloxy bond. |
3.2 The silicate tetrahedron.
At STP oxygen is four-coordinate with Si to form an SiO4 tetrahedron ((SiO44-, e.g., Liebau, 1985; Figure 1). These tetrahedra can then share one or more vertices to form a enormous variety of polymeric structures. By contrast, carbon chemistry is hardly characterized by polymeric oxyanions! Indeed, the only well-characterized carbon oxyanion is carbonate (CO32-); even orthocarbonate (CO44-) has not been isolated, though certain of its esters exist, and polycarbon oxyanions do not exist at all. Presumably the relative weakness of the C-O bond results both from the small size of the C atom and the competing stability of the C=O double bond.
Formally, therefore, silicates can be viewed as salts of various "polysilicic acids," but this proves to be of limited utility because of the extreme degree of polymerization present in many cases. Indeed, attempting such a classification proved to be a blind alley in early silicate investigations. The modern classification is based on the degree of polymerization of the SiO4 tetrahedra, and it is briefly reviewed below.
Interestingly, the fact that 3d hybridization is small apparently favors the extensive polymerization of silicate tetrahedra. The orthophosphate ion (PO43-) is tetrahedral and isoelectronic with SiO44-, but shows relatively little tendency to polymerize; phosphate groups will share two vertices to make polyphosphate anions such as those in ADP/ATP, but molecular P4O10, with 3 shared vertices, is the maximum. Furthermore, the P-O-P bonds are high-energy, which is why ATP-ADP is such a useful energy-storage couple in biosystems. Purely phosphatic 2D or 3D structures do not exist, and under normal conditions P will not even substitute to any significant degree for Si in highly polymerized silicates. Ab initio studies indicate that 2p-3d hybridization in PO43- is considerably stronger than in SiO44- (O'Keeffe et al., 1985), and bridging oxygens do not contribute to it very well. So phosphate "avoids" multiple P-O-P bonds (Yarif, 1987).
In summary, the readiness of Si and O to polymerize to form structures linked by disiloxy bonds, as well as the strength and directionality of the Si-O bond due to its partly covalent character, are the most important characteristics for MNT. Although C-C bonds dominate in carbon macromolecules, disiloxy bonds dominate in Si-bearing macromolecules and fundamentally characterize silicates as well as related compounds such as siloxanes. These considerations, as well as the sheer abundance of Si and O, thus motivate investigating a molecular nanotechnology, not of silicon itself, but of silicates. Furthermore, the stability of C=C double bonds and of delocalized aromatic bonding is a reason that carbon-based macromolecules do not tend to build 3D frameworks, because of the relative instability of tetrahedral "diamondoid" bonding. Hence it may well be that a silicate-based MNT will also be easier to achieve than one based on diamondoid structures.
3.3 Silicate Structures
3.3.1 Isolated anions.
Nesosilicates (neso = "island") or "orthosilicates" are not polymerized at all, but contain isolated SiO44- tetrahedra which thus are formally analogous to such simple oxyanions as phosphate or sulfate. An important example is olivine, (Mg,Fe)2SiO4, which makes up much of the Earth's mantle and is a locally important mineral even at the surface. Although ionic crystals do not seem to offer advantages for nanotechnology (Drexler, 1992, p. 255), they may be useful for resources as they are more easily broken up, as discussed below. Similarly, silicates with small polymeric oxyanions (sorosilicates or oligosilicates) seem not to offer particular advantages for nanotechnology.
The formula for olivine underscores another fundamental characteristic of silicate chemistry: cations of similar charge and size (in this case, Fe2+ and Mg2+) can substitute freely into the structure. Hence compositions can show wide variation within a constant structure type.
3.3.2 Chains.
Silicates can also polymerize into infinite chain structures (inosilicates), which include the pyroxenes and pyroxenoids (single chains; Figure 2a), the amphiboles (double chains; Figure 2b), and more complex structures, such as the "tube silicates" mentioned below (Liebau, 1985). Single-chain structures have the empirical formula MSiO3 and were often called "metasilicates" in the older literature, a term that lingers in industrial usage but that should be obsolete. In natural minerals, these chains are ionically bound by interstitial metal ions such as Mg2+, Fe2+, or Al3+.
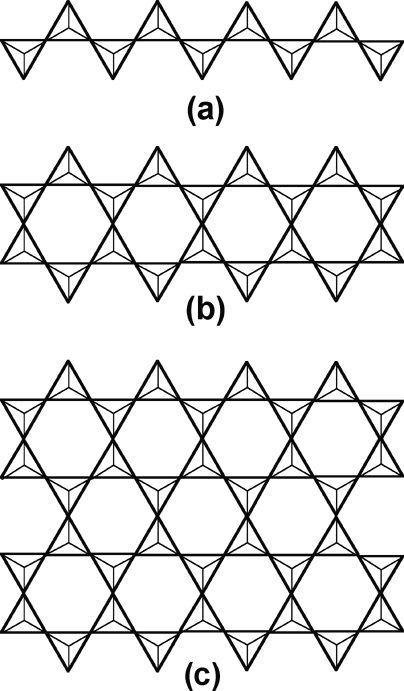 |
| Figure 2. Silicate structures. (a) Chain structure in pyroxenes. (b) Double chain in amphiboles. The structures in (a) and (b) extend indefinitely both right and left. (c) Sheet structure in mica and clay minerals. This structure extends indefinitely in two dimensions. Individual tetrahedra as in Figure 1 are shown diagrammatically; where tetrahedra are joined at a vertex, the oxygen is shared by the vertices. |
Cyclic structures (cyclosilicates), in which a chain is linked end-to-end to form a ring, also occur; beryl, Be3Al2[Si6O18], with an Si6O18 ring, is a naturally occurring example. Cyclosilicates have been synthesized with ring sizes up to Si12O36 (Liebau, 1985, p. 99, 193) Rings with the number of silicons having factors of 2, 3, or 4 are favored, although Winkler & Hoebbel (1989) reported the synthesis of a 7-ring [Si7O21] cyclosilicate.
Although cyclic and chain structures are still in part ionically bound, as they are polymeric oxyanions, certain structures have properties that may motivate their being goals of MNT synthesis. Certain promising solid electrolytes are based on an [Si12O36] ring structure (Shannon et al., 1978). Chain silicates may also make useful fibers; e.g., one form of asbestos (tremolite) is an amphibole. Of course, many fibrous silicates are currently a source of concern as potential carcinogens. However, the carcinogenicity results from the inhalation of tiny fibers. The nanotechnological fabrication of defect-free, macroscopic fibers might be expected not only to be considerably stronger, as the macroscopic strength would be limited not by defects but by the Si-O bond strength, but also considerably safer as they could not be inhaled.
3.3.3 Sheets.
The sharing of 3 tetrahedral vertices leads to infinite sheet structures (phyllosilicates; phyllo = "leaf"). Important phyllosilicates include the micas and clay minerals, both of which are abundant as minerals. These are also first natural silicate structures in which 4-coordinate Al substituting for Si becomes important. Al-substitution leaves a net negative charge to the sheet, which is compensated by included cations that also hold the sheets together.
The silicate sheet in these minerals consists of adjacent hexagonal rings of tetrahedra with their unshared apex all pointing to the same side of the sheet (Liebau, 1985; see also Bailey, 1984 for a detailed discussion of the mica structure) (Figure 2c). The empirical formula of this anionic sheet is Si2O5-2. (In the old literature what are now known to be sheet structures were commonly termed "disilicates." This terminology is archaic and must be avoided, especially as some modern authors use "disilicate" to describe the Si2O7-6 ion; i.e., a double tetrahedral oligomer.) Other sheet structures exist; anthophyllite, with alternating 4 and 8-membered rings, occurs naturally, and even more sheet structures have been crystallized artificially (Liebau, 1985).
Although silicate sheet structures may have little to offer a mature nanotechnology, they may help with the intermediate steps. Cleaved mica sheets are one of the easiest ways of obtaining atomically flat surfaces of macroscopic (~cm2) size (e.g., Bragg et al., 1965, pp. 259; Tabor & Winterton, 1969; Claesson et al., 1986; Chen et al., 1992), and this has motivated their extensive use in scanning-force microscopy, commonly as a substrate for an Au coating (e.g., DeRose et al., 1991; Hegner et al., 1993). The cleavage face of a mica consist of the hexagonal side of the silicate sheet; K+ ions in the hexagonal sites bind the uncleaved crystal together, and it is the weakness of this electrostatic bond that accounts both for the ease and perfection of the cleavage.
3.4 Siloxanes.
Siloxanes ("silicones") can be considered as silicate chain, sheet, or oligomeric structures in which the oxygen unused for a polymeric disiloxy bond has been replaced with hydrogen or an organic side chain. As is well known, organosiloxane polymers have many useful properties that can be varied with the degree of polymerization and the nature of the side chains: they are quite stable, inert to most reagents, and have low vapor pressures, which result both from the great strength and stability of the Si-O bond and that of the Si-C bond (e.g., Noll, 1968; Table 1).
Polyhedral siloxanes in which each silicon is linked to three others via disiloxy bonds are termed silsesquioxanes (sesquisiloxanes) (Barry et al., 1955). Strictly speaking, the atom ratio of O:Si should be 3:2 (sesqui- = "one and a half") (e.g., Rikowski & Marsmass, 1993); however, "silsesquioxane" also seems to be used more broadly for any oligomeric polyhedral siloxane (Feher et al., 1989). Silsesquioxanes sensu stricto are topologically equivalent to a sphere and are commonly called spherosiloxanes (e.g., Agaskar & Klemperer, 1995).
Siloxane diversity is shown by the spherosiloxane "cubosiloxane," H8Si8O12 (Bornhauser & Calzaferri, 1990), which can be considered the siloxy analog of cubane (C8H8) in which each C-C bond is replaced by an Si-O-Si bond (Figure 3). Cubosiloxane, moreover, is both considerably easier to synthesize and more stable than cubane (Agaskar, 1991). In passing, formal replacement of C-C linkages by Si-O-Si could lead to vast numbers of new Si-based structures.
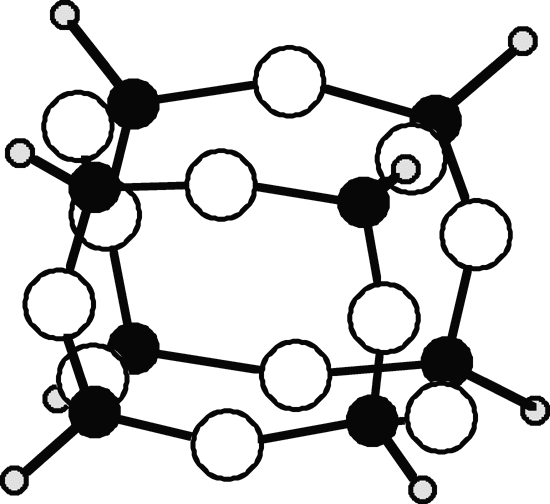 |
| Figure 3. Structure of the spherosiloxane "cubosiloxane", H8Si8O12. Large white circles are O atoms; medium black circles Si, small shaded circles H.
|
The close relation between siloxanes and silicates is underscored by work such as that of Feher and coworkers (Feher et al., 1989; Feher & Budzichowski, 1995), who have used substituted silsesquioxanes as models for silica surfaces for understanding metal coordination during catalysis. Siloxanes also show how organic chains could be attached to a silicate "backbone," and silicate and diamondoid structures thus be combined. Finally, as described in the following paper, modern sol-gel processes for glass and crystalline silicate synthesis commonly use silicon alkoxides (alkoxysilanes and alkoxysiloxanes) as starting materials. These compounds have general formulas SiR4 (alkoxysilane) or SiOR2 (alkoxysiloxane), where R is an alkoxy group (OCH3, OC2H5, etc.) Formally, these compounds can be considered derivatives of silane (if no disiloxy link is present) or siloxanes; alternatively, they can be considered esters of orthosilicic acid (Si(OH)4, a protonated silicate tetrahedron) or polymeric silicic acids, respectively.
Although the disiloxy link is formally analogous to an ether link, it is considerably more polar so that it is both more hydrophilic and more susceptible to hydrolysis. Hence, siloxanes are not much like ethers, and cyclosiloxanes are generally not good complexing agents, in contrast to crown ethers. Nonetheless, a handful of cyclosiloxane complexes have been reported (Churchill et al., 1993; Eaborn et al., 1995).
3.5 Aqueous chemistry of silicates.
As emphasized previously, silicates will spontaneously polymerize into 3D structures out of aqueous solution, in contrast to sp3 carbon. This is promising for silicate nanotechnological assembly: not only may ambient-temperature conditions be used, without such engineering difficulties as high vacuum, but assembly directly out of aqueous solution may be practical. Hence, the aqueous chemistry of silicates will be briefly reviewed.
At pH values less than ~8, Si(OH)4 is the dominant species in aqueous solutions of silica at low total Si concentrations (< ~10-3 M). At higher concentrations the silanol (Si-O-H) groups spontaneously polymerize to yield higher oligomers linked by a disiloxy bond (e.g., Iler, 1979, Ch. 3; Dent Glasser & Lachowski, 1980ab; Andersson et al., 1982):
-Si-O-H + Si-O-H ==> -Si-O-Si- + H2O.
Reaction is most favorable when one of the silanols is deprotonated to an Si-O- group (Iler, 1979, p. 355). These oligomers grow into colloid-sized silica particles by Ostwald ripening, in which larger particles grow at the expense of smaller ones (Iler, 1979, Ch. 3; Brinker & Scherer, 1990, pp. 99-107; Bergna, 1994a) to form a sol. Ultimately these colloids themselves become crosslinked, and once the whole volume is crosslinked, the sol has become a gel. Water is still present, but only interstitially in an interconnected 3D polymeric network (Iler, 1979, Ch. 5; Andersson et al., 1982). Gelation occurs most rapidly at moderately acidic pH (5-6); it is slowest at the silica isoelectric point (pH ~2), probably because the Si(OH)4 monomers are all fully protonated. At even lower pH, polymerization evidently is catalyzed by H+ (Iler, 1979, p. 257).
At higher pH (>8), however, much more concentrated silicate solutions are stable (e.g., Iler, 1979; Dent Glasser & Lachowski, 1980ab). In part this is because disiloxy bonds undergo nucleophilic attack by OH-:
- Si - O - Si + OH- ==> Si - O- + Si - OH,
evidently via a 5-coordinate intermediate (e.g., Liebau, 1984; Holmes, 1990; Kubicki et al., 1993). (Note again the contrast to carbon chemistry.) This is also the mechanism by which silicates dissolve in basic solutions. In part, however, the sols are stabilized by mutual electrostatic repulsion; the colloidal particles acquire a negative charge through partial deprotonation, because silanol groups on large polymeric units are much more acidic (Iler, 1979, p. 182-185). Indeed, Iler (1979, p. 129) emphasizes the bimodal distribution of silicate species in such sols, with monomers and small oligomers in quasi-equilibrium with the colloidal particles. Such systems are in a sense 2-phase (Iler, 1982).
At high Si:Na ratios (> ~1), small oligomeric silica species become significant, as described more fully in the next paper. In compositions typical of conventional "sodium silicate" solutions (2.1  [Si]tot 9.3; 2.4 [Na+]tot 9.6), for example, Svensson et al. (1986) inferred the presence of dimers, trimers, and tetramers, including cyclic species; with Si/Na ~ 3.3, at the threshold of gelation, cagelike species predominate. These inferences were based on 29Si NMR. [Si]tot 9.3; 2.4 [Na+]tot 9.6), for example, Svensson et al. (1986) inferred the presence of dimers, trimers, and tetramers, including cyclic species; with Si/Na ~ 3.3, at the threshold of gelation, cagelike species predominate. These inferences were based on 29Si NMR.
Although under equilibrium conditions the degree of silica that can be dissolved is limited even at high pH, high silica sols (>4:1 Na:Si) can be metastable at lower pH (8-10). Such sols can undergo little change over timescales of months and even years, because electrostatic repulsion and (probably) mutual steric hindrance inhibits the crosslinking of the colloidal particles (Healy, 1994). Metastability is achieved by techniques for increasing the size of the colloidal particles, which inhibits the gelling kinetics (Iler, 1979, Ch. 4); traces of additives that change the colloid surface chemistry have also been used (Iler, 1979, p. 323).
As described in the following paper, such silica sols are the raw material for many traditional silicate syntheses, especially zeolites, and also find use in ceramic synthesis. However, silica sols are at best statistical mixtures, containing a hodgepodge of silicate oligomers, polymers, and colloids that reflect some compromise of thermodynamics and kinetics (Iler, 1979, p. 161-163; Dent Glasser & Lachowski, 1980ab; Falcone, 1982). This limits their utility, particularly in modern synthetic procedures that aspire to better control over the synthesis at the molecular level. To be attractive for MNT, a means would need to be found to select out only particular oligomers with the desired size and shape. This issue is discussed further in the following paper.
4. Silicate Chemistry, II. Framework Structures.
4.1 Tectosilicates.
Sharing all four vertices of the SiO4 tetrahedron yields an infinite 3D tetrahedral framework (a "T-framework"), a tectosilicate. A fully polymerized 3D network of SiO4 tetrahedra simply has the stoichiometry SiO2, and indeed silica occurs in a number of such frameworks. (Since each oxygen is shared with another silicon, the overall stoichiometry is SiO(4/2) = SiO2.) The polymorphs of 4-coordinate SiO2 that have thermodynamic stability fields (quartz, cristobalite, keatite, tridymite, and coesite) have such 3D structures (Heaney, 1994); however, these structures are sufficiently compact that molecular-sized voids do not exist. Several silica clathrasils (defined below) have also been synthesized, and more open but metastable SiO2 frameworks have been synthesized artificially (Higgins, 1994). Silicalite, which is zeolite ZSM-5 in which no Al is present (e.g., Szostak, 1989, p. 5), is one example.
The unstrained disiloxy bond angle is ~140°, and many natural silicates have angles near this value (Liebau, 1985, pp. 24-30). Nonetheless, bending energies of the disiloxy bond are low from about 120° to 180° (Gibbs & Boisen, 1986; Boisen et al., 1994), although the bond strongly resists bending to sharper angles. Indeed, Bieniok & Bürgi (1994), in a normal-mode analysis of a small silicate "building unit" in various crystal and molecular structures, found that 95% of the distortion from the block's ideal symmetry could be modeled by rigid tetrahedra hinged at their vertices. This low bending energy leads to an extraordinary number of possible T-framework structures, as the bond angle can be opened out considerably without straining the bonds unduly. Indeed, the small energy differences among the 4-coordinate silica polymorphs results merely from the different bending of the Si-O-Si bonds (Navrotsky, 1994). In addition, substitution of other atoms for Si in the tetrahedral framework, as described below, also can lead to bonds with angles  140°. This results from the size and electrostatic repulsion of included cations, and from steric effects due to polymerization and to "guest" molecules incorporated during crystallization 140°. This results from the size and electrostatic repulsion of included cations, and from steric effects due to polymerization and to "guest" molecules incorporated during crystallization
The large literature on the topology of possible tetrahedral frameworks is surveyed briefly in the following paper. As might be expected, however, many more frameworks can be theoretically described than can evidently be achieved under the limitations of present-day "shake and bake" synthesis techniques (Brunner, 1990). It is noteworthy that unknown 3D silica structures have been found by simulated annealing models (Boisen et al., 1994; Teter et al., 1995). This suggests that, at least in part, kinetic factors determine which silica structures actually crystallize. This is yet another indication of the flexibility of the disiloxy bond and its potential nanotechnological applications, and a nanotechnological "assembler" approach to synthesis should make a vastly greater number of networks achievable.
4.2 T-Cation substitution and aluminosilicates.
Though even all-silica frameworks would have multiple uses, the variety of tectosilicates is greatly broadened because other 4-coordinate cations can be substituted for Si in the tetrahedral framework. Indeed, in general one can speak of a "T" (tetrahedral) atom when describing possible framework structures. In natural systems, tetrahedral aluminum substitution, up to an Al/Si ratio of ~1:1, is ubiquitous. Indeed, the feldspars, one group of alumino-tectosilicates with general formula (Xx,Y1-x)(Six,Al1-x)AlSi2O8 (X = Na, K; Y = Ca), are the most abundant compounds in the Earth's crust.
Al substitution leaves a net negative charge on the framework, which then must be compensated by included cations; e.g., in anorthite feldspar, CaAl2Si2O8, the aluminum atoms are part of the T-framework, and the included Ca2+ maintains charge balance. As Al avoids Al-O-Al bonds ("Loewenstein's rule"; Loewenstein, 1954), Al substitution generally does not occur beyond a ratio of 1:1.
Probably the most interesting aluminosilicates for nanotechnological applications are zeolites, which were mentioned as the prototypes of open framework structures. Zeolites are members of the broad class of compounds called molecular sieves (Szostak, 1989, p. 2-4); sensu stricto, they are framework aluminosilicates that support internal voids at least large enough to accommodate water molecules, and that also are interconnected via channels sufficiently wide that the molecules can be exchanged with the environment (Smith, 1988). (Void-bearing structures in which the voids are isolated by passages too small to accommodate molecules are called clathrasils (Liebau, 1985).) Thus, non-aluminosilicate molecular sieves are not truly zeolites, though the term is often used more broadly, especially for pure silica structures. Some zeolites occur naturally as minerals; a great many others, however, have been synthesized artificially and are of great technological importance, especially in catalysis.
(Ironically, Drexler (1992, p. 254-255) implied that aluminum will become unimportant in the future as structural metals become obsolescent. This may be true for Al metal, but Al might still have major importance in MNT as a component of aluminosilicate frameworks. The sheer abundance of Al is also a motivation for its use.)
Other substitutions of the T cation are possible and vastly broaden the possibilities. Many 4-coordinate atoms, including III-V elements (B, P, Ga, As), transition metals (Fe, Co, Ti, Mn, Zn), and even mono-and divalent cations such as Li and Be have been substituted into T-frameworks (e.g., Flanigen et al., 1986; Flanigen, 1991; Szostak, 1991; Feng et al., 1997ab). Even in natural systems, a decision as to when a 4-coordinate cation is a member of the tetrahedral framework is somewhat arbitrary; e.g., beryl is traditionally described as based on [Si6O18]-12 anion rings, but Be is 4-coordinate in the structure and can alternatively be described as participating with Si in a 3D T-framework in which the Be and Si sites are ordered (Zoltai, 1960). The same is true of Be in phenacite, Be2SiO4, and Zn in willemite, Zn2SiO4.
Thus, the chemical properties of the T-framework can be tailored by both variations in the linkage of its tetrahedral structure, and by the chemical microenvironment induced by the tetrahedral atoms. However, destabilizing the framework could be a concern: For example, Hansen (1990) suggested that framework voids could be widened arbitrarily by insertion of BeO42- tetrahedra between the SiO4 tetrahedra. The Be-O-Si bond is considerably less flexible than the disiloxy bond, however, and has an equilibrium angle of only ~129° (Downs & Gibbs, 1981); hence such structures may be vulnerable to collapse as the bonds would be strongly strained.
Substitution of P into a silicate framework merits a brief note. Artioli et al. (1984) and Lok et al. (1984) reported the synthesis of silicon-bearing aluminophosphates, but these still have electrically negative or neutral frameworks as (on an atom basis) Al  P. P strongly prefers to substitute for Al rather than Si (Derouane et al., 1990), and indeed P substitution for Si in natural aluminosilicates is negligible (Yarif, 1987). Nonetheless, Dyer et al. (1987) have successfully synthesized a P-bearing framework by brute-force methods (soaking zeolite crystals in a phosphate melt at ~230°C!). This yields a cationic T-framework in which the charge compensation must occur by included anions. Most immediately, as these authors note, such a framework might lead to an anion-selective ion exchanger, and it could have many further applications such as in the ion sorter/separator described above. P. P strongly prefers to substitute for Al rather than Si (Derouane et al., 1990), and indeed P substitution for Si in natural aluminosilicates is negligible (Yarif, 1987). Nonetheless, Dyer et al. (1987) have successfully synthesized a P-bearing framework by brute-force methods (soaking zeolite crystals in a phosphate melt at ~230°C!). This yields a cationic T-framework in which the charge compensation must occur by included anions. Most immediately, as these authors note, such a framework might lead to an anion-selective ion exchanger, and it could have many further applications such as in the ion sorter/separator described above.
A molecular nanoassembler probably could assemble a P-bearing framework without such extreme conditions. Despite the avoidance of PO4 tetrahedra for sites in a fully polymerized T-framework under the normal conditions of chemical synthesis, PO4 tetrahedra could presumably be included by a nanoassembler that inserted tetrahedra atomistically, as the energy difference of PO4 vs. other tetrahedral cations is, after all, relatively small (Derouane et al., 1990).
4.3 Silicate-like structures.
Silicon is not the only element that forms giant-molecule anions and frameworks with oxygen; germanates, as might be expected, are largely homologous, and have been used to model silicate structural changes with pressure for decades, as they undergo analogous changes at much lower pressure (Ringwood & Seabrook, 1963). Combinations of elements introduce particular variety: for example, although phosphorus alone is limited in its ability to form complex structures with oxygen, a wide variety of "hybrids" with other elements exists. Aluminum phosphate, AlPO4, which is isoelectronic with SiO2, has a similar giant-molecule structure and even has analogous polymorphs; the one with the a-quartz structure occurs naturally as the rare mineral berlinite. In effect, all Si-O-Si links are replaced by Al-O-P. P also forms complex framework and sheet structures with certain transition and actinide elements (e.g., W, Mo, U, Zr, V), and some of these are comparable to zeolites (Corbridge, 1995, p. 275-295).
Hence, it is hardly surprising that completely non-silicate molecular sieves exist, and AlPO4 sieves, several of which are isostructural with known zeolites, have been a particular research focus (e.g., Flanigen et al., 1986; Wilson et al., 1982). A number of other non-silicate molecular sieves been synthesized recently (Flanigen, 1991), such as the gallophosphate "cloverite" (Estermann et al., 1991), which has large internal voids, and zinco(beryllo) phosphate and arsenate frameworks (Gier & Stucky, 1991).
Recently, giant-molecule anions with N instead of O as the linking electronegative atom have been synthesized. "Nitridometallates" (Niewa & Jacobs, 1996), based on polymerized tetrahedra MN4 (M = W, Mo, Nb, Ta) have structures related to silicates. "Nitridosilicates" (e.g., Huppertz & Schnick, 1996), which are based on a SiN4-8 tetrahedron, and "nitridophosphates" (Schnick, 1993), based on a PN4-12 tetrahedron, can be even more complex, as more than two tetrahedra can share a vertex (Huppertz & Schnick, 1996). Their synthesis, however, is difficult due to the stability of N2; it could be easier with a nanotechnological approach, though the possible utility of these compounds remains unknown.
Among the most interesting non-silicates are borates, which are more complex than silicates because B can occur in either trigonal (BO3) or tetrahedral (BO4) coordination with oxygen. Indeed, BO3 and BO4 groups are commonly linked to form complex anions in the same crystal structure (e.g., Christ & Clark, 1977); evidently the energy difference between the coordination states is very small and depends on subtle interactions with the neighbor atoms. Moreover, borate molecular sieves containing both 3- and 4-coordinate B exist and have unusual bulk properties (e.g., Ghose, 1982). Cationic B-Al bearing frameworks have also been synthesized recently (Yu et al., 1994, 1996ab). B atoms can even change their coordination with hydration state (e.g., Sauer, 1992), as in H-boralite, a synthetic zeolite with the ZSM-5 structure in which Al has been replaced by B in the T-sites (Schoole & Veeman, 1985; quoted in Sauer, 1992). Such a change in coordination, and hence structure, resulting from changes in the ambient concentration of particular molecules may prove useful in MNT systems.
Nonetheless, although borate and borosilicate chemistry are receiving increased attention, B is a very rare element. Not only has this reduced research into borate structures to an esoteric specialty, it suggests that any MNT applications will probably be limited to specialized purposes.
4.4 Structural variations.
Borates also show that more exotic linkages than perfect 3D T-frameworks are not only possible but potentially useful. Other cations not in T-coordination, such as octahedrally coordinated cations (Flanigen et al., 1986) might be incorporated into the framework as long as their bonding is not purely ionic. In addition, a handful of silicates have 3D structures in which not all tetrahedral vertices are shared (an "interrupted framework"; Liebau, 1985, p. 243-244). Stacking defects in zeolitic structures, or sites where a T-cation (usually Al) has been replaced by 4 OH terminations on the coordinating oxygens, show other alternatives. Of course, whatever their potential utility, with present synthesis techniques there is no atomistic control over such framework modifications.
4.5 Silicate glasses.
These are of obvious importance and merit a brief mention. Such glasses are highly polymerized, with the degree of polymerization rising with the SiO2 content. Stoichiometric SiO2 glasses are obviously fully polymerized, and so although tough and corrosion-resistant have high softening temperatures. Commercial soda glass contains some Al substituting for Si and alkali cations to balance the charge; this yields lower working temperatures but also lowered strength and corrosion resistance. An enormous variety of cations can be introduced for various purposes (e.g., Pb for "crystal" glass), and the polymeric framework can also be varied, e.g., by introducing B instead of Al.
All such materials, being glasses, have no long-range atomic order, although the nanoenvironment of Si is largely the same as in crystals; nearly all are 4-coordinate with O. "Interrupted" frameworks can occur, too, in which a few tetrahedral vertices are not shared but are instead terminated with F or OH; these include such natural amorphous silicas as opal and those precipitated by certain organisms.
5. Strength of Silicates
A major motivation for MNT development is the promise of extreme-strength materials; with no defects or grain boundaries at which cracks can form and propagate, the strengths of atomically perfect materials should approach the limits set by chemical bonds. This motivation is likely to be especially important in the near term development of MNT, as such materials, having no moving parts, seem easier to fabricate than nanomechanical systems (Gillett, 1996a).
Hence the ultimate strength of silicates is of considerable interest. For simplicity, unsubstituted silicate structures (i.e., pure silica) only will be considered here. Not only do such structures intuitively seem stronger, because of the absence of ionic repulsion effects, but a net framework charge would need to be compensated by included ions, which would greatly complicate modeling and also add mass to the structure.
Obviously, the tensile strength of silica structures is dominated by the strength of the Si-O bond. A simple estimate from Young's modulus and the atomic spacing of undeformed quartz yields a value of 16 GPa (Kelly & MacMillan, 1986, p. 6). A more sophisticated estimate, by Nàray-Szabó & Ladik (1960; quoted in Kelly & MacMillan, 1986, p. 8), yields a force of roughly 2 x 10-9 N to dissociate one O from an SiO4 tetrahedron. The force-field model of Hill & Sauer (1994) yields a very similar value of ~2.2 x 10-9 N to dissociate a single Si-O bond. Obviously, the strength of macroscopic silicates then depends on the density of bonds per unit area; using an estimate of 8 x 1018 bonds/m2 in silica glass (Doremus, 1973; quoted in Kelly & MacMillan, 1986, p. 8) yields a strength of ~15.9 GPa. All these estimates compare well with an experimental maximum strength, measured on silica fibers under nearly ideal conditions, of ~15 GPa (Kelly & MacMillan, 1986, p. 371).
Diamond-based materials, with a specific mass/bond value less than half that of silica and theoretical tensile strengths > 100 GPa, are clearly much stronger, at least in theory. Nonetheless, the silica value is comparable to the estimated theoretical strengths of other strong solids (Kelly & MacMillan, 1986, p.6), and is at least several times as strong as the strongest conventional materials. In addition, silica has no cleavage plane, in contrast to diamond, and this may compensate somewhat for the difference in strength.
6. Silicates as Resources.
No discussion would be complete without underscoring the sheer abundance of silicates. Si and O constitute, respectively, 20.5 and 60.4 atom percent of the Earth's crust (e.g., Mason, 1966; Figure 4). Nearly all rock-forming minerals are silicates, and silicates thus underlie most of geochemistry. However, aside from bulk uses such as gravel, fill, and so on, silicates have been not been traditionally a focus in economic geology: the sheer strength of the Si-O bond has made common rock unattractive as a source of raw material. Instead, most ore minerals are the relatively rare non-silicates, chiefly sulfides or oxides, and less commonly carbonates or sulfates.
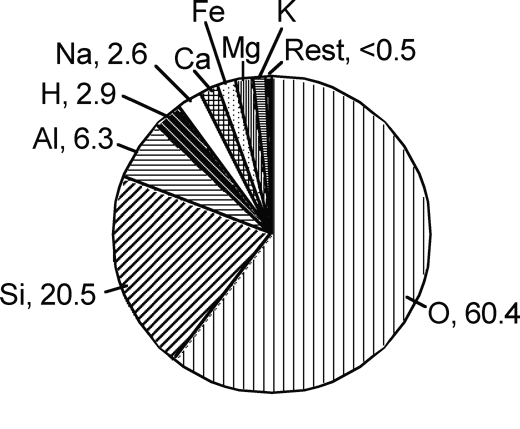 |
| Figure 4. Crustal abundances of the Earth, in mole fraction. The reason for the dominance of aluminosilicates in ordinary rock is strikingly evident. (Data calculated from Mason, 1966.)
|
6.1 Extraction: silicate sols.
Although potential silicate raw materials are literally everywhere, not all are equally easy to get. It is often noted that common sand (which is mostly composed of grains of quartz) is an obvious feedstock, as it is used today as the ore for Si. However, the attractiveness of quartz sand as an Si (or silicate) resource merely reflects the limitations of current bulk technological extraction techniques, in which a feedstock must be as enriched in the desired element as possible because of the heat-intensive nature of conventional extraction processes (Gillett, 1996a). Essentially pure quartz sand is concentrated naturally by sedimentary processes, and this purity is the deciding economic factor.
With more mature extraction capabilities sand would be far less attractive. Quartz crystals are extremely hard and have no cleavage: i.e., they do not tend to break along particular crystallographic planes. In fact, just these properties make it a passable abrasive. Hence, crushing and grinding quartz is both expensive and tough on machinery.
Given a mature separation technology, fine-grained silicate and aluminosilicate materials, in the size range of clay (<~4 µm) to silt (<~ 30 µm) would be far more attractive. Indeed, clay and silt are extremely abundant at the Earth's surface, occurring sometimes in kilometers-thick deposits (e.g., deep-water lake and marine sediments). These materials are made up both of silicates comminuted by mechanical weathering (chiefly quartz and feldspars), and clay minerals, the latter commonly being weathering products as they are precipitated during alteration of other silicates. Table 2 gives analyses of some representative "grab samples" of fine-grained "dirt" from various localities in northwestern Nevada; the sheer abundance of SiO2 in ordinary Earth materials should be noted.
Table 2. Analyses of grab samples of miscellaneous "dirt" and mine tailings from selected locations in northwestern Nevada.
| |
|
BRP |
|
OF |
|
SP |
|
TLD |
|
YT |
|
MT |
| Major elements (oxide percentages by weight): |
| SiO2 |
|
43.1 % |
|
66.0 % |
|
52.5 % |
|
57.5 % |
|
64.2 % |
|
67.6 % |
| TiO2 |
|
00.49 |
|
00.67 |
|
00.70 |
|
00.74 |
|
00.47 |
|
00.51 |
| Al2O3 |
|
11.8 |
|
15.0 |
|
16.3 |
|
17.5 |
|
14.6 |
|
13.2 |
| Fe2O3 |
|
05.32 |
|
03.86 |
|
06.98 |
|
06.03 |
|
04.79 |
|
03.18 |
| MnO |
|
00.08 |
|
00.12 |
|
00.14 |
|
00.14 |
|
00.00 |
|
00.00 |
| MgO |
|
03.46 |
|
01.02 |
|
03.13 |
|
02.32 |
|
00.96 |
|
00.61 |
| CaO |
|
04.51 |
|
02.12 |
|
02.33 |
|
04.31 |
|
01.31 |
|
00.33 |
| Na2O |
|
02.29 |
|
02.55 |
|
02.01 |
|
03.14 |
|
02.71 |
|
01.30 |
| K2O |
|
02.82 |
|
02.91 |
|
02.53 |
|
02.06 |
|
02.81 |
|
03.25 |
| P2O5 |
|
00.19 |
|
00.16 |
|
00.29 |
|
00.21 |
|
00.20 |
|
00.05 |
| LOI |
|
25.4 |
|
05.92 |
|
13.4 |
|
05.22 |
|
06.33 |
|
08.57 |
| Total |
|
99.52 |
|
100.31 |
|
100.23 |
|
99.18 |
|
98.40 |
|
98.64 |
| Selected minor elements (parts per million, by weight): |
| Ba |
|
0763 ppm |
|
1192 ppm |
|
0885 ppm |
|
1060 ppm |
|
0935 ppm |
|
1309 ppm |
| Co |
|
0014 |
|
0011 |
|
0022 |
|
0017 |
|
0006 |
|
0002 |
| Cr |
|
0064 |
|
0118 |
|
0054 |
|
0086 |
|
0261 |
|
0037 |
| Ga |
|
0020 |
|
0016 |
|
0021 |
|
0028 |
|
0019 |
|
0023 |
| Ga |
|
0020 |
|
0016 |
|
0021 |
|
0028 |
|
0019 |
|
0023 |
| Nb |
|
0010 |
|
0017 |
|
0015 |
|
0007 |
|
0014 |
|
0034 |
| Ni |
|
0037 |
|
0018 |
|
0038 |
|
00021 |
|
0017 |
|
0003 |
| Pb |
|
0013 |
|
0013 |
|
0017 |
|
00010 |
|
00nd |
|
0017 |
| Rb |
|
0121 |
|
0105 |
|
0101 |
|
0073 |
|
0076 |
|
0084 |
| Sr |
|
0354 |
|
0361 |
|
0365 |
|
0539 |
|
0428 |
|
0049 |
| V |
|
0143 |
|
0102 |
|
0143 |
|
0163 |
|
0081 |
|
0033 |
| Y |
|
0032 |
|
0032 |
|
0027 |
|
0026 |
|
0018 |
|
0055 |
| Zn |
|
0148 |
|
0073 |
|
0130 |
|
0090 |
|
0021 |
|
0049 |
| Zr |
|
0161 |
|
0276 |
|
0168 |
|
0129 |
|
0136 |
|
0530 |
| Cu |
|
0042 |
|
0024 |
|
0046 |
|
0035 |
|
0543 |
|
0002 |
| Semiquantitative mineral compositions: |
| Quartz (SiO2) |
|
Mj |
|
Mj |
|
Mj |
|
Mj |
|
Mj |
|
Mj |
| Feldspars |
|
Mj |
|
Mj |
|
Mj |
|
Mj |
|
Mj |
|
Mn |
| Micas |
|
Mj |
|
|
|
|
|
Mn |
|
Mn |
|
|
| Clay minerals |
|
|
|
Mn |
|
Mj |
|
Mn |
|
Tr |
|
Mn |
| Pyroxenes |
|
|
|
Mj |
|
|
|
|
|
|
|
|
| Amphiboles |
|
|
|
|
|
|
|
Mn |
|
|
|
|
| Non-silicates |
|
Mn |
|
|
|
|
|
|
|
Mn |
|
Mn |
Localities: BRP, Black Rock Playa (i.e., dry lake bed), Pershing Co.; OF, Owyhee Flat, Elko Co.; SP, Stagecoach Playa, Lyon Co.; TLD, Lake Lahontan deltaic deposit, Truckee Canyon, Washoe Co. These four samples all have the consistency of clay or fine mud (grain size of clay to fine silt, corresponding to diameters of less than a few micrometers). MT, tailings from vicinity of Midas, Elko Co.; YT, tailings from old copper smelter near Yerington, Lyon Co. The grain size of these latter two samples ranges from clay to fine sand.
Analyses of major elements (those common in the Earth's crust) are conventionally reported in terms of oxides. Oxygen is by far the dominant anion in the crust, but is not usually analyzed for directly. Instead, the elements are assumed to be bound to oxygen. "LOI" is "loss on ignition", which is material that is volatilized and driven off below ~100°C. It consists of organic matter and loosely bound water. The analyses do not sum to 100% largely because of analytical imprecision; other elements are highly unlikely to be present at more than ppm levels. Note the overwhelming abundance of silica in ordinary Earth materials.
A semiquantitative tabulation of mineral abundances, based on X-ray powder data, is also given. "Mj" = major component; "Mn" = minor component; "Tr" = trace. "Feldspars" includes both alkali feldspars (albite and potassium feldspars) and plagioclase (Ca-Na feldspar). "Micas" includes sheet silicates of probable detrital origin, including muscovite and clinochlore. "Clay minerals" include sheet silicates of probable authigenic origin (i.e., precipitated during weathering processes). Pyroxenes and amphiboles (mostly hornblende) are chain silicates of probable detrital origin. "Non-silicates" include calcite (CaCO3), gypsum (CaSO4·2H2O), and jarosite (KFe3(OH)6(SO4)2); not all are present in any given sample.
Samples collected by author; analyses courtesy of P. Lechler, Nevada Bureau of Mines and Geology, Reno, Nevada.
Obviously, comminution of such material would not be required. Conventional crushing and grinding not only leads to high maintenance costs, it is extremely expensive energetically, as little of the applied energy goes into generating new surface energy. Most is wasted as heat (Gordon et al., 1987, p. 44).
Solubilizing such silicates and aluminosilicates is relatively easy because of their small grain sizes. Indeed, the solubility of clay minerals is a problem in conventional Al extractive metallurgy (Bayer process) because the dissolved silica is then difficult to remove from the caustic liquor (e.g., Roach & White, 1987; Bánvölgi et al., 1990); hence conventional aluminum ores must be as silica-free as possible. However, given nanotechnological developments such as atomically precise, highly specific semipermeable membranes, for which there seem to be strong economic drivers already (Gillett, 1996a), separating such a solution should be relatively straightforward.
Another possibility is solubilizing coarser-grained but unpolymeric silicates (i.e., nesosilicates) in strong mineral acids, such as HCl. Whether a silicate "gelatinized" (i.e., formed a silica gel, by polymerization of released SiO4 groups) on acid treatment was once a standard test in determinative mineralogy. Soon after the true nature of silicate structures was resolved, Murata (1943) showed that silicates that gelatinize either have isolated silicate anions (nesosilicates), or are aluminotectosilicates with Al:Si >2:3. With no disiloxy bonds to break, a nesosilicate is much more easily attacked by hydrogen ions than are polymeric structures. (A high percentage of aluminum in aluminous tectosilicates, however, also makes the framework vulnerable to attack, as Al-O-Si links are readily cleaved by acid.) Indeed, during World War II a process for using olivine as an ore of Mg was developed (Houston, 1949); silica gels were an unwanted byproduct (!). Whether this source proves attractive may depend on whether the Mg and Fe byproducts are useful. In addition, ferroan slags, such as those from copper and steel making, also contain nesosilicates.
Obviously, the product of such dissolution processes will ultimately be a silicate sol. Conventionally, however, aqueous sodium silicate is prepared by fusing a mixture of quartz sand and sodium carbonate at high temperature (1100-1400°C) and dissolving the product in water. Such an energy-intensive procedure is again required with current technology because the purity of SiO2 as quartz sand is the overriding consideration.
6.2 Mining waste and silicate MNT.
Considering slags as resources also suggests an interesting synergy of environmental considerations with silicate raw materials: exploiting the silicate debris left by conventional mining and smelting operations. This would not merely minimize impact, but help ameliorate old impacts, as the waste from conventional mining operations consists largely of enormous quantities of more or less comminuted silicate debris (i.e., crushed rock). Moreover, slags are largely ferroan silicate glasses.
As mentioned, because of the high energy required to break Si-O bonds, silicates are seldom attractive as ore minerals; instead, they nearly always comprise the bulk of the "gangue," the valueless minerals that must be separated from the ore minerals. For example, copper ore from a so-called "porphyry copper" deposit, which currently supply about 50% of the world's Cu (Gordon et al., 1987), typically consists of less than 1% of copper sulfides dispersed through a granite matrix (an igneous rock consisting largely of coarse-grained quartz and feldspars). Such ore rock must be crushed and ground ("beneficiated") merely to separate out the ore minerals; hence, not only is the energy needed to break the rock up mechanically been largely wasted, but the very finely comminuted waste from such mineral dressing operations (the "tailings") presents a serious disposal problem. Such material should furnish an excellent feedstock for silicate-based MNT, however, especially as it will also be relatively reactive due to its comminution. Table 2 also gives analyses of two tailings samples; one in particular (YT) comes from several square miles of tailings around an abandoned porphyry copper mine in western Nevada.
To digress with a personal observation: to one who has spent much of his career associated with the natural resource business, one of the most exciting things about MNT is its implication that all the traditional notions about what constitutes a "resource" have become obsolete. When the very waste left by mining becomes a valuable resource, obviously paradigms have changed beyond recognition.
6.3 Carbothermal extraction of elemental Si.
Elemental Si is at present obtained by the high-temperature carbothermal reduction of silica:
SiO2 + 2 C ==> 2 CO + Si, T ~1500°C.
Obviously this is extremely energy-intensive. To be sure, in cases where reduced Si is required, a minimum expenditure of energy to cleave Si-O bonds is demanded by the laws of thermodynamics. However, siloxanes and other partly or wholly oxidized Si-based compounds are also conventionally prepared from elemental Si. The only important exceptions are bulk construction materials, glasses, and the sodium silicate sols described above, and synthesis of the latter two materials is also energy-intensive. Synthetic approaches for other Si compounds largely begin with oxidation of elemental Si by chlorine or chlorocarbons to yield highly reactive silicon chlorides, where the highly reactive Si-Cl bond is used as the starting point for further syntheses (e.g., hydrolysis to form siloxane polymers) (e.g., Kendrick et al., 1989). Even the silicon alkoxides, which are increasingly used in "sol-gel" synthesis of silicates, are conventionally manufactured from silicon chloride ultimately derived from carbothermal Si.
Obviously such synthesis paths are needlessly energy intensive. Particularly when the ultimate synthetic goal is itself a silicate, it is wasteful to reduce and then re-oxidize Si. Indeed, this expense would largely negate the advantages of silicates' abundance. Milder routes to silicate building blocks are thus highly desirable.
6.4 Extraction: chimie douce approaches.
Several groups have demonstrated low-temperature chimie douce ("soft chemistry") routes to complex silicates and organosilicon compounds directly from simple inorganic silicates at ambient temperatures. Such approaches, which avoid the highly energy-intensive step of carbothermic reduction of SiO2, are also obviously attractive on environmental grounds. They have not yet been looked at in the context of extractive metallurgy, however, and in particular their possible application to feedstocks like the mining waste described above has not been investigated.
It has long been known that catechol (1,2 dihydroxybenzene; Figure 5, I) is an excellent complexing agent for Si, and a basic catechol solution will even dissolve quartz (Bach & Sticher, 1966) to yield tris(catecholato) silicate, M2[o-C6H4O2]3Si, where M is a unipositive cation. (Note that this compound has 6-coordinated Si.) Boudin et al. (1988) synthesized substituted silanes by the action of organolithium or Grignard reagents on this complex, and Ray et al. (1991) used it as a starting point for catecholato-silicate polymers. Boudin et al. (1989) emphasize that the reactivity of the hypercoordinate Si in the catecholate is particularly attractive for such syntheses. Another group (Perry & Lu, 1992; Harrison & Loton, 1995) has investigated the decomplexation of catecholates as a route for preparing amorphous silica. Substituted catechols are also effective complexing agents (Iler, 1977), as is the tropolone anion (Figure 5, II) in acidic or neutral solution (Muetterties & Wright, 1964; Sjöberg et al., 1985). Both may indeed be involved in the biological mechanisms that handle silica (Iler, 1977; Sjöberg et al., 1985). However, tropolone complexes have apparently been made only from silica sols; the dissolution of solid silicates evidently has not been investigated.
 |
| Figure 5. Silicon complexing agents. I, catecholate anion; II, tropolonate anion. |
The "trimethylsilylation" method (Lentz, 1964) has attracted interest as a method of preparing polymers directly from inorganic silicates (Currell et al., 1974; Hefter & Kenney, 1982). Under acidic "scarce water" conditions, the unshared oxygens of a silicate oligomeric oxyanion will react with chlorotrimethylsilane:
-Si-O- + SiCl(CH3)3 + H+ ==> -Si-O-Si(CH3)3 + HCl,
and this generally occurs with minimal disruption of the structure of the oxyanion. For this reason trimethylsilylation has also become a standard technique in silicate study, as the trimethylsilyl-substituted groups can be separated and determined with gas chromatography (e.g., Currell et al., 1984).
As trimethylsilylation requires chlorotrimethylsilane, however, it does not avoid completely the necessity for a reduced silicon compound. More promisingly, Goodwin & Kenney (1988, 1989, 1990; Kenney & Goodwin, 1988) subsequently obtained silicon alkoxides directly from various inorganic silicates by their dissolution and consequent esterification in anhydrous acidified alcohols. Furthermore, the resulting esters largely reflect the structure of the original silicate anion; hence, the syntheses also provide unexpected control over the products obtained. In particular, Harrington & Kenney (1992) synthesized a complex chain siloxane directly from K2CuSi4O10, a "tube" chain silicate, with preservation of the complex silicate "backbone." These alkoxides can be used directly in sol-gel synthesis, or further reduced to siloxanes by Grignard reagents.
Finally, Laine et al. (1991) showed that even quartz sand will dissolve in a solution of NaOH and ethylene glycol to yield glycolates (e.g., Na2[C2H4O2]5Si2), which can in turn be used for sol-gel or hydrothermal syntheses of various ceramics (Kansal & Laine, 1994, 1995) or even of zeolites (Herreros et al., 1994; Herreros & Klinowski, 1995). These complexes contain 5-coordinate Si and are even more reactive than most hypercoordinate Si species, a further advantage when used as bases for syntheses.
7. Silicates and Space Development
A silicate-based nanotechnology with the capability of using common rock as raw material may also be relevant to using local resources on other Solar System bodies, which is commonly thought necessary to make extensive off-Earth activities affordable (e.g., Lewis et al., 1993). The Moon, for example, is essentially devoid of both water and organic matter, except possibly at the poles (Feldman et al., 1998). This obviously is a major obstacle in devising scenarios in which a lunar base could utilize local resources to minimize resupply from Earth (e.g., Heiken et al., 1991), and equally obviously a carbon-based MNT would be of limited help. However, the lunar regolith, the fragmented layer of "soil" covering the lunar surface, consists largely of silicate debris comminuted by eons of meteoritic bombardment. A silicate MNT for exploiting silicate mine waste would require little adaptation to such a feedstock (Gillett, 1997).
8. Sources of Data on Silicates
One reason silicate chemistry is not so well known to nonspecialists as is, say, organic chemistry is that its literature is fragmented. For historical reasons, it is dispersed among mineralogists, petrologists, ceramicists and glass scientists, industrial chemists (especially for zeolites and other molecular sieves), and inorganic chemists; even organic chemists who work on siloxanes can be involved.
Silicate chemistry also was not understood until the second quarter of the 20th century. Nineteenth-century attempts to interpret silicates in terms of salts of "ortho", "meta", and so forth "silicic acids", which had proven reasonably successful in interpreting the oxysalts of elements such as sulfur and phosphorus, went nowhere. (The confusion prevailing on the eve of x-ray methods may be gleaned from the interpretations in Clark (1914).) In retrospect this is not surprising, both because of the giant-molecule polymeric structures of silicate anions, and because the identity of the included cations in silicate minerals is not important, merely their size and charge. Only in 1930 did Bragg (1930), the pioneer of the atomistic determination of crystal structures by x-ray diffraction, propose a systematic description of silicate structures based on the degree of polymerization of SiO4 tetrahedra. A minor modification of his classification system is still used today (Liebau, 1985). Berman (1937) tentatively extended Bragg's classification to then-known minerals, but the modern knowledge was not incorporated into standard mineralogy texts until after World War II. Even now, silicate terminology is not completely standardized (Liebau's (1985) suggestions are the most comprehensive), and it seems not quite integrated into the mainstream of inorganic chemistry.
Terminology has no doubt provided another barrier for outsiders. Mineralogists use an opaque hodgepodge of arbitrary names for minerals; although this has some conveniences, because of the compositional variability of natural materials, it does not lend itself to a systematic nomenclature. Zeolite terminology is even worse, consisting of a mishmash of both mineral names and arbitrary labels conferred by industrial and research chemists.
This history contrasts with organic chemistry, whose roots go well back into the 19th century, and which was systematized at a much earlier date. Biochemistry, furthermore, seems never to have become so estranged from organic chemistry as had mineralogy from inorganic chemistry.
A second reason that silicates have been something of a "backwater" is probably their propensity to form giant molecule anionic structures rather than isolated molecules. Thus they do not lend themselves to the sort of step-by-step synthesis of isolated, albeit complicated, molecules that characterize traditional organic chemistry. Rather, sets of empirical "recipes" to encourage preferential crystallization of one or another structure have been developed. To be sure, similar empirical reaction rules pervade organic chemistry as well; but nearly all such rules have reasonable atomistic rationalizations that have proven very useful, not only in explaining known reactions, but in predicting synthesis pathways. Such synthesis reactions can also be strung together indefinitely to make desired organic molecules, and, of course, a major part of the art of organic synthesis is devising such stepwise syntheses. Crystallization "recipes" are much less conducive to such an approach.
Even more than in most nanotechnological endeavors, then, an interdisciplinary approach is required. Hence this brief overview on sources of information about silicates. Obviously in the space available this can merely note literature sources that may be useful besides the obvious sources in the major chemistry journals.
8.1 Mineralogy.
The mineralogy literature is obviously directed toward natural systems, with a major focus being the significance of compositional variations, both as a guide to the pressure-temperature conditions of mineral formation, and their effects on the details of the silicate structure. Hence thermochemical data is another major focus; Berman (1988) has compiled and recalculated an internally self-consistent set of thermochemical data for the oxide components of geologically important silicates, and Robie and Hemingway (1995) have recently published a new revision of their thermochemical data compilation relevant to minerals.
(Thermochemical data in the geological and ceramics literature is commonly expressed with oxides as the reference state. A silicate can be treated formally as a combination of oxides; e.g., Mg2SiO4 = 2 MgO·SiO2. This convention is convenient because the differences in energies between the formal oxide components and the resulting silicate is much smaller than is that between the component elements and oxygen, so that the energy differences between alternative silicates are not swamped by uncertainties in forming the oxides from the elements. The convention can, however, be a source of serious confusion.)
Finally, in recent years there has been a growing emphasis on predictive rather than descriptive modeling of silicate structures, including ab initio studies.
Important journals include American Mineralogist, Mineralogical Magazine, and The Physics and Chemistry of Minerals. Much relevant material is also in Clays and Clay Minerals and Clay Mineralogy. The Mineralogical Society of America's Reviews of Mineralogy series includes particularly valuable compendia on various mineral groups. Finally, important mineral groups have been the subjects of treatises, as with feldspars (Smith & Brown, 1988).
8.2 Ceramics and glasses.
A number of journals cover this area (e.g., Journal of Non-Crystalline Solids; Physics and Chemistry of Glasses; Glass and Ceramics), and it obviously grades into the voluminous materials science literature. The classic volumes Phase Diagrams for Ceramists (Levin et al., 1964, 1969, 1975; Roth et al., 1981, 1983) also contain much valuable information on silicate thermochemistry.
Of particular interest is the Materials Research Society's Better Ceramics through Chemistry series (Brinker et al., 1984, 1986, 1988; Cheetham et al., 1994; Coltrain et al., 1996; Hampden-Smith et al., 1992; Zelinski et al., 1990), which contains a great deal of information on the synthesis, structure, and modeling of ceramic materials, including silicates. Sol-gel processing, especially approaches based on alkoxide hydrolysis, is a particular focus of many papers.
Slags, another waste product from conventional extractive metallurgy, are themselves largely silicate glasses, but information on them is largely found in the metallurgical literature, such as the Transactions of the AIME. As noted above, slags may find unexpected value as resources.
8.3 Zeolites.
Zeolites have an enormous literature. Recent books and review papers include Barrer (1982), Catlow (1992), Dyer (1988), Meier (1986), Rabo (1976), Smith (1988), and Szostak (1989). Zeolite research is spread over two groups, natural zeolites (mineralogy) and synthetics (industrial chemistry), but there seems to be much communication between the two, and indeed, a modest amount of mining of natural zeolites for industrial use takes place even now. A journal (Zeolites) is devoted to them, and International Zeolite Conferences are held every few years, which typically furnish the subject of a proceedings volume (e.g., van Santen & Jacobs, 1989).
8.4 Aqueous silicates and sols.
This is also a somewhat separate subfield in which there is a great deal of industrial interest because of the commercial importance of sodium silicate solutions. Many academic studies appear in the standard chemistry journals, although there is some overlap with the mineralogy literature, especially for silica precipitation. Water-soluble silicates are likely to be of particular interest for silicate MNT for two reasons. First, they are potentially a source of silicate "building blocks", as is discussed further in the following paper. Second, as mentioned, silicates conceivably can be assembled directly out of aqueous solution, in complete contrast to sp3 carbon structures. As described above, silicates readily precipitate 3D structures from aqueous solution, and the nanotechnological problem becomes guiding and controlling that polymerization at a molecular level.
The comprehensive source for information up to the late 1970s is Iler's (1979) classic treatise. This came out, however, just before 29Si NMR spectroscopy became widespread, and such studies have vastly clarified our knowledge of the silica species in solution; some of this early work is covered in the 1982 ACS volume (Falcone, 1982). A recent ACS volume on colloidal silica (Bergna, 1994b), a review paper (Swaddle et al., 1994) and several recent research papers (Kinrade et al., 1996, 1998ab) provide entries into the newer literature.
9. Conclusions.
Although for historical reasons silicate chemistry is not so familiar as is that of carbon, silicate-based structures seem to be an attractive alternative for MNT. The sheer abundance of silicates provide one motivation, and the strength of the Si-O bond also compares favorably with other ultra-strong materials. In addition, silicates' thermal and chemical stability, the readiness with which they can form 3D structures containing molecule-sized voids (with or without a net framework charge), and their probable relative ease of synthesis compared to diamondoid structures are also attractive. The last feature is likely to prove particularly important in near-term, embryonic applications of MNT.
Acknowledgments
I thank Bob Mackey for reading an early draft of this paper. I am also grateful to Jim Lewis for putting an html version online in the archives for the Fifth Conference on Molecular Nanotechnology (http://www.foresight.org/Conferences/MNT05/Nano5.html).
References Cited.
- Agaskar P A 1991 New synthetic route to the hydridospherosiloxanes Oh-H8Si8O12 and D5h-H10Si10O15, Inorg. Chem. 30 2707
- Agaskar P A and Klemperer W G 1995 The higher hydridospherosiloxanes: synthesis and structures of HnSinO1.5n; n = 12,14,16,18, Inorg. Chim. Acta 229 355
- Andersson K R, Dent Glasser L S & Smith D N 1982 Polymerization and colloid formation in silicate solutions, in Soluble Silicates (ACS Symposium Series 194) ed J S Falcone p 115-132
- Anpo M, Yamashita H, Ichihashi Y, Fujii Y & Honda M 1997 Photocatalytic reduction of CO2 with H2O on titanium oxides anchored within micropores of zeolites: Effects of the structure of the active sites and the addition of Pt, J. Phys. Chem. B 101 2632-2636
- Artioli G, Pluth J J & Smith J V 1984 Synthetic phosphorus-substituted analcime, Na3Al24Si13P11O96·16H2O with ordered Al and Si/P, Acta Cryst.C40 214-217
- Bach R & Sticher H 1966 Abbau von Quarz mit Brenzkatechin, Experientia 22 515-516
- Bailey S W 1984 Classification and structure of the micas, in Micas (Reviews in Mineralogy 13) ed S W Bailey (Mineralogical Society of America) p 1-12
- Bánvölgi G, Toth A C & Tassy I 1990 In situ formation of sodium aluminum hydrosilicate from kaolinite, in Light Metals 1991 ed E L Rooy (AIME) p 5-15
- Bard A W, & Mallouk T 1992 Electrodes modified with clays, zeolites, and related microporous solids, in Molecular Design of Electrode Surfaces ed R W Murray (Wiley) p 271-312
- Barrer R M, Hydrothermal Chemistry of Zeolites Academic 1982
- Barry A J, Daudt W H, Domicone J J, Gilkey J W 1955 Crystalline organosilsesquioxanes, J. Am. Chem. Soc. 77 4248-4252
- Barton T J, Boudjouk P 1990 Organosilicon chemistry: A brief overview, in Silicon-Based Polymer Science: A Comprehensive Resource (Adv. Chem. 224), eds J M Zeigler & F W G Fearon (American Chemical Society) p 3-46
- Bergna H E 1994a Colloid chemistry of silica: an overview, in The Colloid Chemistry of Silica (Adv. Chem. 234) ed H E Bergna (American Chemical Society) p 1-50.
- Bergna H E, ed 1994b The Colloid Chemistry of Silica (Adv. Chem. 234) American Chemical Society
- Bein T & Enzel P, 1993 Inclusion of conducting polymers in inorganic hosts: toward conducting nanostructures, in Intrinsically Conducting Polymers: An Emerging Technology, ed M Aldassi (Kluwer Academic) p 51-60
- Berman H 1937 Constitution and classification of the natural silicates, Am. Mineral. 22 342-408
- Berman R G 1988 Internally-consistent thermodynamic data for minerals in the system Na2O-K2O-CaO-MgO-FeO-Fe2O3-Al2O3-SiO2-TiO2-H2O-CO2, J.Petrology 29 445-522
- Bieniok A M & Buergi H-B, 1994 Structural distortions in Si8O12 fragments and D4R units: a comparison between silsesquioxane molecules and LTA zeolites, J. Phys. Chem. 98 10735-41
- Boisen M B Jr, Gibbs G V & Bukowinski, M S T 1994 Framework silica structures generated using simulated annealing with a potential energy function based on an H6Si2O7 molecule, Phys. Chem. Min. 21 269-284
- Boudin A, Cerveau G, Chuit C, Corriu R J P & Reye C 1988 Reactivity of dianionic hexacoordinated silicon complexes toward nucleophiles: A new route to organosilanes from silica, Organometallics 7 1165-1171
- Boudin A, Cerveau G, Chuit C, Corriu R J P & Reye C 1989 Reactivity of hypervalent silicon derivatives. One step synthesis of mono- and dihydrosilanes, J. Organomet. Chem. 362 265-72
- Bornhauser P & Calzaferri G 1990 Normal coordinate analysis of H8Si8O12, Spect. Acta 46A 1045-1056
- Bragg W L 1930 XXV. The structure of silicates, Z. Krist. 74 237-305
- Bragg L, Claringbull G F & Taylor W H 1965 Crystal Structures of Minerals (Ithaca NY: Cornell Univ. Press)
- Brinker C J, Clark D E & Ulrich D R, eds 1984 Better Ceramics Through Chemistry (Materials Res. Soc. Symp. Proc. 32)
- Brinker C J, Clark D E & Ulrich D R, eds 1986 Better Ceramics Through Chemistry II (Materials Res. Soc. Symp. Proc. 73)
- Brinker C J, Clark D E & Ulrich D R, eds 1988 Better Ceramics Through Chemistry III (Materials Res. Soc. Symp. Proc. 121)
- Brinker C J & Scherer G W 1990 Sol-Gel Science: The Physics and Chemistry of Sol-Gel Processing (Academic Press)
- Brunner G O 1990 Criteria for the evaluation of hypothetical zeolite frameworks, Zeolites 10 612
- Cano M L, Cozens F L, Fornes V, Garcia H & Scaiano J C 1996 Intrazeolite photochemistry. 12. ship-in-a-bottle synthesis and control of the photophysical properties of 9-(4-methoxyphenyl)xanthenium ion imprisoned into large-pore zeolites, J. Phys. Chem. 100 18145-18151
- Catlow C R A ed 1992 Modelling of Structure and Reactivity in Zeolites (Academic Press)
- Cheetham A K, et al eds 1994 Better Ceramics Through Chemistry VI (Materials Res. Soc. Symp. Proc. 346)
- Chen Y L, Chen, S Frank C & Israelachvili J 1992 Molecular mechanism and kinetics during the self-assembly of surfactant layers, J. Coll. Interf. Sci. 153 244-265
- Christ C L & Clark J R 1977 A crystal-chemical classification of borates with emphasis on hydrated borates, Phys. Chem. Min. 2 59-87
- Chuit C, Corriu R J P, Reye C & Young J C 1993 Reactivity of penta- and hexacoordinate silicon compounds and their role as reaction intermediates, Chem. Rev. 93 1371-??
- Churchill M R, Lake C H, Chao S H L & Beachley O T Jr 1993 Silicone grease as a precursor to a pseudo crown ether ligand: crystal struture of [K+]3[K(Me2SiO)7+][InH(Ch2CMe3)3-]4, Chem. Comm. 1577
- Claesson P M, Herder P, Stenius P, Eriksson J C & Pashley R M 1986 An ESCA and AES study of ion-exchange on the basal plane of mica, J. Coll/ Interf. Sci. 109 31-39
- Clark F W 1914 The constitution of the natural silicates, USGS Bull. 588
- Collins S J, & O'Malley P J 1995 A theoretical description for the monomolecular cracking of C-C bonds over acidic zeolites, J. Catal. 153 94-99
- Coltrain B K et al. eds 1994 Better Ceramics Through Chemistry VII, Organic/Inorganic Hybrid Materials (Materials Res. Soc. Symp. Proc. 435)
- Corbridge D E C 1995 Phosphorus, an Outline of its Chemistry, Biochemistry, and Uses, 5th ed Elsevier
- Cruickshank D W J 1961 The role of 3d orbitals in p bonds between (a) silicon, phosphorus, sulphur or chlorine and (b) oxygen or nitrogen, J. Chem. Soc. 5486-5504
- Cruickshank D W J 1985 A reassessment of dp-pp bonding in the tetrahedral oxanions of second-row atoms, J. Mol. Struct. 130 177-191
- Currell B R, Midgley H G, Seaborne M A & Thakur C P 1974 Preparation of polyorganosiloxane from mineral silicates using the method of trimethylsilylation, Br. Polym. J. 6 229-240
- Currell B R, Midgley H G, Mingham B J, Parsonage J R, Vidgeon E A, 1984 The use of trimethylsilylation to study the structure of mineral silicates, JCS Dalton Trans. 5 757-760
- De Almeida W B & O'Malley P J 1991 The potential energy surface of H4SiO4, Chem. Phys. Lett. 178 483-487
- De Almeida W B & O'Malley P J 1992 An ab initio molecular orbital study of the potential energy surface for the orthosilicate anion, [H3SiO4]-, J. Mol. Struct. (Theochem) 258 379-387
- De Almeida W B & O'Malley P J 1992 Ab initio molecular orbital calculated stationary points on the potential energy surface of [H3AlO4]---similarities with H4SiO4, J. Mol. Struct. (Theochem) 257 305-312
- Dent Glasser L S 1979 Non-existent silicates, Z. Krist. 149 291-305
- Dent-Glasser L S & Lachowski E E 1980a Silicate species in solution. Part 1. Experimental observations, JCS Dalton Trans. 393-398
- Dent-Glasser L S & Lachowski E E, 1980b Silicate species in solution. Part 2. The structure of polymeric species, JCS Dalton Trans. 399-402
- DeRose J A, Thundat T, Nagahara L A & Lindsay S M 1991 Gold grown epitaxially on mica: conditions for large area flat faces, Surf. Sci. 256 102-108
- Derouane E G, Fripiat J G & von Ballmoos R 1990 Quantum mechanical calculations on molecular sieves. 2. Model cluster investigation of silicoaluminophosphates, J. Phys. Chem. 94 1687-1692
- Downs J W & Gibbs G V 1981 The role of the BeOSi bond in the structures of beryllosilicate minerals, Am. Mineral. 66 819-826
- Drexler K E 1992 Nanosystems Wiley
- Dyer A, Malik S A, Araya A & McConville T J 1987 New anion exchangers of zeolitic type, in Recent Developments in Ion Exchange, eds P A Williams & M J Hudson (Elsevier) p 257-263
- Dyer A 1988 An Introduction to Zeolite Molecular Sieves, John Wiley & Sons
- Eaborn C, Hitchcock P B, Izod K & Smith J D, 1995 Two diorganopotassates: crystal structure of (K(C6H6))(K(C(SiMe3)2(SiMe2Ph))2), Angew. Chem. Int. Ed. Engl., 34 2679
- Estermann M, McCusker L B, Baerlocher C, Merrouche A & Kessler H 1991 A synthetic gallophosphate molecular sieve with a 20-tetrahedral-atom pore opening, Nature 352 320-323
- Falcone J S ed 1982 Soluble Silicates (Am. Chem. Soc. Symp. Series 194)
- Feher F J, Budzichowski T A 1995 Silasesquioxanes as ligands in inorganic and organometallic chemistry, Polyhedron 14 3239
- Feher F J, Newman D A & Walzer J F 1989 Silsesquioxanes as models for silica surfaces, J. Am. Chem. Soc., 111 1741-8
- Feldman W C, Maurice S, Binder A B, Barraclough B L, Elphic R C & Lawrence D J 1998 Fluxes of fast and epithermal neutrons from Lunar Prospector: Evidence for water ice at the lunar poles, Science 281 1496
- Feng P, Bu X & Stucky G D 1997a Hydrothermal syntheses and structural characterization of zeolite analog compounds based on cobalt phosphate, Nature 388 735-741
- Feng P, Bu X, Tolbert S H & Stucky G D 1997b Syntheses and characterizations of chiral tetrahedral cobalt phosphates with zeolite ABW and related frameworks, J. Am. Chem. Soc., 119 2497-2504
- Finger L W & Hazen R M 1991 Crystal chemistry of six-coordinated silicon: A key to understanding the Earth's deep interior, Acta Cryst. B47 561-580
- Flanigen E M 1991 Zeolites and molecular sieves. An historical perspective, in Introduction to Zeolite Science and Practice, eds H van Bekkum, E M Flanigen & J C Jansen (Elsevier) p 13-34
- Flanigen E M, Lok B M, Patton R L & Wilson S T 1986 Aluminophosphate molecular sieves and the periodic table, Pure Appl. Chem. 58 1351-1358
- Ghose S 1982 Stereoisomerism of the pentaborate polyanion [B5O12](9-), polymorphism and piezoelectricity in the hilgardite group of minerals, a novel class of polar borate zeolites, Am. Mineral., 67 1265-1272
- Gibbs G V & Boisen M B Jr 1986 Molecular mimicry of structure and electron density distributions in minerals, Mat. Res. Soc. Symp. Proc. 73 515-527
- Gibbs G V, Boisen M B Jr, Downs R T & Lasaga A C 1988 Mathematical modeling of the structures and bulk moduli of TX2 quartz and cristobalite structure-types, T=C, Si, Ge and X=O, S, Mat. Res. Soc. Symp. Proc. 121 155-65
- Gibbs G V, Downs J W & Boisen M B Jr 1994 The elusive SiO bond, in Silica; Physical Behavior, Geochemistry and Materials Applications (Reviews in Mineralogy 29) eds P J Heaney, C T Prewitt & G V Gibbs (Mineralogical Society of America) p 331-368
- Gier T E & Stucky G D 1991 Low-temperature synthesis of hydrated zinco(beryllo)-phosphate and arsenate molecular sieves, Nature 349 508-510
- Gillett S L 1996a Nanotechnology, pollution control, and resources, Nanotechnology 7 177-182
- Gillett S L 1996b Near-term nanotechnology: The molecular fabrication of nanostructured materials, Nanotechnology 7 168-176
- Gillett S L 1997 Implications of molecular nanotechnology for space resources, in Space Manufacturing 11 ed B Faughnan (American Institute of Aeronautics and Astronautics) p 115-123
- Goodwin G B & Kenney M E 1988 A new approach to the synthesis of alkyl silicates and organosiloxanes, in Inorganic and Organometallic Polymers: Macromolecules containing Silicon, Phosphorus, and Other Inorganic Elements (Am. Chem. Soc. Symp. Series 360) eds M Zeldin, K J Wynne & H R Allcock p 238-248,
- Goodwin G B & Kenney M E 1989 Novel route to Ca8Si4O12Cl8, alkoxycyclotetrasiloxanes, aryloxycyclotetrasiloxanes, alkylcyclotetrasiloxanes and arylcyclotetrasiloxanes from wollastonite (CaSiO3) US patent 4,824,985
- Goodwin G B & M E Kenney 1990 A new route to alkoxysilanes and alkoxysiloxanes of use for the preparation of ceramics by the sol-gel technique, Inorg. Chem. 29 1216-??
- Gordon R B, Koopmans T C, Nordhaus W D & Skinner B J 1987 Toward a New Iron Age? Harvard Univ. Press
- Haile S M, Wuensch B J, Laudise R A, Siegrist T & Foxman B M 1993 Crystallography and composition of some new potassium-neodymium silicates, in The Structural Chemistry of Silicates (Trans. Am. Cryst. Assoc. 27) ed A C Wright p 77-93
- Hampden-Smith M J, Klemperer W G & Brinker C J eds 1992 Better Ceramics Through Chemistry V (Materials Res. Soc. Symp. Proc. 271)
- Hansen S 1990 Expanding zeolite-type nets, Nature 346 799-800
- Harrington B A & Kenney M E 1992 The preparation and properties of a fibrous tube alkoxysiloxane derived from a tube silicate, Coll. Surf. 63 139
- Harrison C C & Loton N 1995 Novel routes to designer silicas: studies of the decomposition of (M+)2[Si(C6H4O2)3]·xH2O. Importance of M+ identity of the kinetics of oligomerization and the structural characteristics of the silicas produced, JCS Faraday Trans. 91 4287-97
- Healey T W 1994 Stability of aqueous silica sols, in The Colloid Chemistry of Silica (Adv. Chem. 234) ed H E Bergna (American Chemical Society) p 147-159
- Heaney P J 1994 Structure and chemistry of the low-pressure silica polymorphs, in Silica: Physical Behavior, Geochemistry and Materials Applications (Reviews in Mineralogy 29) eds P J Heaney, C T Prewitt & G V Gibbs (Mineralogical Society of America) p 1-40
- Hefter J & Kenney M E 1982 Synthesis of the tube silicate litidionite and structural relationships between it and some other silicates, Inorg. Chem. 21 2810-16
- Hegner M, Wagner P & Semenza G 1993 Ultralarge atomically flat template-stripped Au surfaces for scanning probe microscopy, Surf. Sci. 291 39-46
- Heiken G, Vaniman D T & French B M 1991 Lunar Sourcebook, Cambridge Univ. Press
- Herreros B & Klinowski J 1995 Hydrothermal synthesis of zeolites from 5-coordinate silicon compounds, J. Phys. Chem. 99 1025
- Herreros B, Carr S W & Klinowski J 1994 5-Coordinate Si compounds as intermediates in the synthesis of silicates in nonaqueous media, Science 263 1585
- Herron N 1989 Toward Si-based life: Zeolites as enzyme mimics, Chemtech 542-548
- Herron N, Wang Y, Eddy M M, Stucky G D, Cox D E, Moller K & Bein T 1989 Structure and optical properties of CdS superclusters in zeolite hosts, J. Am. Chem. Soc. 111 530-540
- Higgins J B 1994 Silica zeolites and clathrasils, in Silica: Physical Behavior, Geochemistry and Materials Applications (Reviews in Mineralogy 29) eds P J Heaney, C T Prewitt & G V Gibbs (Mineralogical Society of America) 507-544
- Hill J-R & Sauer J 1994 Molecular mechanics potential for silica and zeolite catalysts based on ab initio calculations. 1. Dense and microporous silica, J. Phys. Chem. 98 1238
- Holmes R R, 1990 The stereochemistry of nucleophilic substitution at tetracoordinated silicon, Chem. Rev. 90 17-??
- Houston E D 1949 Magnesium from olivine, Trans. AIME 182 113-126
- Huppertz H & Schnick W 1996 BaYbSi4N7--unexpected structural possibilities in nitridosilicates, Angew. Chem. Int. Ed. Engl. 35 1983
- Iler R K 1977 Hydrogen-bonded complexes of silica with organic compounds, in Biochemistry of Silicon and Related Problems, eds G Bendz & I Lindqvist (Plenum) p 53-75
- Iler R K 1979 The Chemistry of Silica; Solubility, Polymerization, Colloid & Surface Properties & Biochemistry of Silica (New York: John Wiley & Sons, Inc.) 866 p
- Iler R K 1982 Colloidal components in solutions of sodium silicate, in Soluble Silicates (ACS Symposium Series 194) ed J S Falcone p 95-114
- Jacobs P A & Martens J A 1987 Synthesis of high-silica aluminosilicate zeolites (Elsevier)
- Jelinek R, Pines A, Ozkar S & Ozin G A 1991 A study of nanostructure assemblies and guest-host interactions in sodium zeolite-Y using 23Na double-rotation NMR, Nanotechnology 2 182-186
- Kansal P & Laine R M 1994 Pentacoordinate silicon complexes as precursors to silicate glasses and ceramics, J. Amer. Cer. Soc. 77 3292
- Kansal P & Laine R M 1995 Group II tris(glycolato)silicates as precursors to silicate glasses and ceramics, J. Amer. Cer. Soc. 78 529
- Kelemen G, Lortz W & Schoen G 1989 Ionic conductivity of synthetic analcime, sodalite and offretite, J. Mat. Sci. 24 333-338
- Kelly A & N H MacMillan 1986 Strong Solids 3rd ed Clarendon
- Kenney M E & Goodwin G B 1988 Silicate esters and organosilicon compounds, US patent 4,717,773
- Kinrade S D, Marat K & Knight C T G 1996 Longitudinal 29Si nuclear magnetic relaxation in aqueous alkali-metal silicate solutions revisited, J. Phys. Chem., 100 18357-???
- Kinrade S D, Knight C T G, Pole D L & Syvitski R T 1998 Silicon-29 NMR studies of tetraalkylammonium silicate solutions. 1. Equilibria, 29Si chemical shifts, and 29Si relaxation, Inorg. Chem. 37 4272
- Kinrade S D, Knight C T G, Pole D L & Syvitski R T 1998 Silicon-29 NMR studies of tetraalkylammonium silicate solutions. 2. Polymerization kinetics, Inorg. Chem. 37 4278
- Krueger J S, Mayer J E & Mallouk T E 1988 Long-lived light-induced charge separation in a zeolite L-based molecular triad, J. Am. Chem. Soc. 110 8232-8234
- Krummenacker M 1994 Steps towards molecular manufacturing, Chem. Design Autom. News 9 29-39
- Kubicki J D, Xiao Y & Lasaga A C 1993 Theoretical reaction pathways for the formation of [Si(OH)5]- and the deprotonation of orthosilicic acid in basic solution, Geochim. Cosmochim. Acta 57 3847
- Laine R M, Blohowiak K Y, Robinson T R, Hoppe M L, Nardi P, Kampf J & Uhm J 1991 Synthesis of pentacoordinate silicon complexes from SiO2, Nature 353 642-644
- Lentz C W 1964 Silicate minerals as sources of trimethylsilyl silicates and silicate structure analysis of sodium silicate solutions, Inorg. Chem. 3 574-579
- Levin E M, Robbins C R & McMurdie H F 1969 Phase Diagrams for Ceramists Vol 1 2nd ed (American Ceramic Society)
- Levin E M, Robbins C R & McMurdie H F 1969 Phase Diagrams for Ceramists Vol 2 (American Ceramic Society)
- Levin E M, Robbins C R & McMurdie H F 1975 Phase Diagrams for Ceramists Vol 3 (American Ceramic Society)
- Lewis J S, Matthews M S & Guerrieri M L eds 1993 Resources of Near-Earth Space (Tucson, AZ: Univ. of Arizona Press)
- Li Z, Wang C M, Persaud L & Mallouk T E 1988 Electrochemistry of metalloporphyrins and viologens and zeolite Y modified electrodes: Evidence for electron trapping by monomolecular porphyrin layers, J. Phys. Chem. 92 2592-2597
- Liebau F 1984 Pentacoordinate silicon intermediate states during silicate condensation and decondensation. Crystallographic support, Inorg. Chim. Acta 89 1-7
- Liebau F 1985 Structural Chemistry of Silicates (Springer Verlag)
- Lok B M, Messina C A, Patton R L, Gajek R T, Cannan T R & Flanigen E M 1984 Silicoaluminophosphate molecular sieves: Another new class of microporous crystalline inorganic solids, J. Am. Chem. Soc. 106 6092-6093
- Loewenstein W 1954 The distribution of aluminum in the tetrahedra of silicates and aluminates, Am. Mineral. 39 92-95
- Mason B 1966 Principles of Geochemistry 3rd ed Wiley
- McDaniel C V & Maher P K 1976 Zeolite stability and ultrastable zeolites, in Zeolite Chemistry & Catalysis (ACS Monograph 117) ed J A Rabo (American Chemical Society) p 285-331
- Meier W M 1986 Zeolites and zeolite-like materials, Pure Appl. Chem. 58 1323-1328
- Merkle R 1995 Design-ahead for nanotechnology, in Prospects in Nanotechnology, eds. M Krummenacker & J Lewis (Wiley) p 23-52
- Muetterties E L & Wright C M 1964 Chelate chemistry. I. Tropolone and aminotroponimine derivatives of the main-group elements, J. Am. Chem. Soc. 86 5132-5137
- Murata K J 1943 Internal structure of silicate minerals that gelatinize with acid, Am. Mineral. 28 545-562
- Murugesamoorthi K A, Srinivasan S & Appleby A J 1993 Research, development, and demonstration of solid oxide fuel cell systems, in Fuel cell systems, eds L J M J Blomen & M N Mugerwa (Plenum Press)
- Navrotsky A 1994 Thermochemistry of Crystalline and Amorphous Silica, in Silica: Physical Behavior, Geochemistry and Materials Applications (Reviews in Mineralogy 29) eds P J Heaney, C T Prewitt & G V Gibbs (Mineralogical Society of America) p 309-330
- Newsam J M 1986 The zeolite cage structure, Science 231 1093-1099
- Newton M D 1981 Theoretical probes of bonding in the disiloxy group, in Structure and Bonding in Crystals (Academic Press) p 175-193
- Niewa R & Jacobs H 1996 Group V and VI alkali nitridometalates: a growing class of compounds with structures related to silicate chemistry, Chem. Rev. 96 2053
- Noll W 1968 Chemistry and Technology of Silicones (Academic Press)
- O'Keeffe M & McMillan P F 1986 The Si-O-Si force field: ab initio MO calculations, J. Phys. Chem. 90 541-542
- O'Keeffe M, Domenges B & Gibbs G V 1985 Ab initio molecular orbital calculations on phosphates: Comparison with silicates, J. Phys. Chem., 89 2304-2309
- Ozin G A 1992 Nanochemistry: synthesis in diminishing dimensions, Adv. Materials 4 612-649
- Ozin G A, Kuperman A & Stein A 1989 Advanced zeolite materials science, Angew. Chem. Int. Ed. Engl. 28 359-377
- Ozin G A & Özkar S 1992 Intrazeolite topotaxy, Adv. Materials, 4 11-22
- Perry C C & Lu Y 1992 Preparation of silicas from silicon complexes: role of cellulose in polymerization and aggregation control, JCS Faraday Trans. 88 2915-21
- Persaud L, Bard A J, Campion A, Fox M A, Mallouk T E, Webber S E & White J M 1987 Photochemical hydrogen evolution via singlet-state electron-transfer quenching of zinc tetra(N-methyl-4pyridyl)porphyrin cations in a zeolite L based system, J. Am. Chem. Soc. 109 7309-7314
- Pool Robert 1994 The smallest chemical plants, Science 263 1698-1699
- Raabe G & Michl J 1989 Multiple bonds to silicon, in The Chemistry of Organic Silicon Compounds Part 2 eds S Patai & Z Rappoport (Wiley Interscience) p 1015-1142
- Rabo J A 1976 Zeolite Chemistry & Catalysis (ACS Monograph 117)
- Ramamurthy V 1991 Photoprocesses of organic molecules included in zeolites, in Photochemistry in Organized and Constrained Media ed V Ramamurthy (VCH) p 429-494
- Ray D J, Laine R M, Viney C & Robinson T R 1991 Silica as a source of silicon-containing compounds/polymers, Polym. Prepr. 32(3) p 550-1
- Reed A E, Schade C, von Rague Schleyer P, Kamath P V & Chandrasekhar J 1988 Anomeric effects involving silicon centres: Silanols, Chem. Comm. 67-69
- Rickert Hans 1982 Electrochemistry of solids: an introduction (Springer-Verlag)
- Rikowski E, Marsmann H C 1997 Cage-rearrangement of silsesquioxanes, Polyhedron 16 3357
- Ringwood A E & Seabrook M 1963 High-pressure phase transformations in germanate pyroxenes and related compounds, J. Geophys. Res. 68 4601-4609
- Roach G I D & White A J 1987 Dissolution kinetics of kaolin in caustic liquors, in Light Metals 1988 ed L G Boxall (The Metallurgical Society)
- Robie R A & Hemingway B S, 1995 Thermodynamic properties of minerals and related substances at 298.15 K and 1 bar (105 Pascals) pressure and at higher temperatures, USGS Bull. 461
- Rolison D R 1990 Zeolite-modified electrodes and electrode-modified zeolites, Chem. Rev. 90 867-878
- Roth R S, Negas T & Cook L P 1981 Phase Diagrams for Ceramists Vol 4 (American Ceramic Society)
- Roth R S, Negas T & Cook L P 1983 Phase Diagrams for Ceramists Vol 5 (American Ceramic Society)
- Sauer R O 1944 Nomenclature of organosilicon compounds, J. Chem. Ed. 303-305
- Sauer J, 1992 Quantum mechanical studies of zeolites, in Modelling of Structure and Reactivity in Zeolites ed C R A Catlow (Academic) 183-216
- Scamehorn C A & Angell C A 1991 Viscosity-temperature relations and structure in fully polymerized aluminosilicate melts from ion dynamics simulations, Geochim. Cosmochim. Acta 55 721
- Schnick W 1993 Solid-state chemistry with nonmetal nitrides, Angew. Chem. Int. Ed. Engl. 32 806
- Shannon R D, Taylor B E, Gier T E, Chen H-Y & Berzins T 1978 Ionic conductivity in Na5YSi4O12-type silicates, Inorg. Chem. 17 958
- Sjöberg S, Öhman L-O & Ingri N 1985 Equilibrium and structural studies of silicon(IV) and aluminium(III) in aqueous solution. 11. Polysilicate formation in alkaline aqueous solution. A combined potentiometric and 29Si NMR study, Acta Chem. Scand. A 39 93-107
- Smith J V 1988 Topochemistry of zeolites and related materials. 1. Topology and geometry, Chem. Rev. 88 149-182
- Smith J V & Brown W L 1988 Feldspar Minerals 2nd ed (Springer-Verlag)
- Stebbins J F 1991 NMR evidence for five-coordinated silicon in a silicate glass at atmospheric pressure Nature 351 638-639
- Stucky G D 1992 The interface of nanoscale inclusion chemistry, Prog. Inorg. Chem. 40 99-178
- Svensson I L, Sjöberg S & Öhman L-O 1986 Polysilicate equilibria in concentrated sodium silicate solutions, JCS Faraday Trans. I 82 3635-3646
- Swaddle T W, Salerno J & Tregloan P A 1994 Aqueous aluminates, silicates, and aluminosilicates, Chem. Soc. Rev. 23 319
- Szostak R 1989 Molecular Sieves: Principles of Synthesis and Identification (Van Nostrand Reinhold)
- Szostak R 1991 Modified zeolites, in Introduction to Zeolite Science and Practice eds H van Bekkum, E M Flanigen & J C Jansen (Elsevier) p 153-200
- Szostak R 1992 Handbook of molecular sieves (Van Nostrand Reinhold)
- Tabor D & Winterton R H S 1969 The direct measurement of normal and retarded van der Waals forces, Proc. Roy. Soc. A 312 435-450
- Teter D M, Gibbs G V, Boisen M B Jr, Allan D C & Teter M P 1995 First-principles study of several hypothetical silica framework structures, Phys. Rev. B 52 8064
- Thomas J M 1992 (April) Solid acid catalysts, Scientific American p 112
- Townsend R P 1991 Ion exchange in zeolites, in Introduction to Zeolite Science and Practice eds H van Bekkum, E M Flanigen & J C Jansen (Elsevier) p 359-391
- Turro N J & Garcia-Garibay M 1991 Thinking topologically about photochemistry in restricted spaces, in Photochemistry in organized and constrained media ed V Ramamurthy VCH p 1-38
- Wilson S T, Lok B M, Messina C A, Cannan T R & Flanigen E M 1982 Aluminophosphate molecular sieves: A new class of microporous crystalline inorganic solids, J. Am. Chem. Soc. 104 1146-1147
- Winkler A & D Hoebbel 1988 Über ein Strontiumbromidsilicat und den molekularen Aufbau seines Silicatanions, Z. Anorg. Allg. Chem. 562 170-174
- Yarif S 1987 Substitution of silicon by phosphorus in silicates, Chem. Erde 46 1-5
- Yu J, Tu K & Xu R 1994 Synthesis and characterization of a novel microporous boron-aluminum chloride with a cationic framework, in Zeolites and Related Microporous Materials: State of the Art 1994 (Studies in Surface Science and Catalysis 84/a) ed J Weitkamp (Elsevier) p 315-322
- Yu J, Xu R, Chen J & Yue Y 1996 On the crystallisation and nature of the microporous boron-aluminium oxo chloride BAC(10), J. Mat. Chem. 3 465
- Yu J, Xu R, Xu Y & Yue Y 1996 Synthesis and characterization of a boron-aluminum oxochloride, J. Sol. State Chem. 122 200
- Zelinski B J J et al eds 1990 Better Ceramics Through Chemistry IV (Materials Research Society Symp. Proc. 180)
- Zoltai T 1960 Classification of silicates and other minerals with tetrahedral structures Am. Mineral. 45 960-973
|