Distributed
Molecular Modeling
over Very-Low-Bandwidth
Computer Networks
by
Will Ware
14 Washington Avenue
Arlington, MA 02174
http://world.std.com/~wware/
This is a draft paper
for a talk at the
Fifth
Foresight Conference on Molecular Nanotechnology.
The final version has been submitted
for publication in the special Conference issue of Nanotechnology.
CA. Copyright 1997 Will
Ware, all rights reserved.
This page uses the HTML <sup> and <sub>
conventions for superscripts and subscripts. If "103"
looks the same as "103" then your browser does not
support superscripts. If "xi" looks the
same as "xi" then your browser does not support
subscripts. Failure to support superscripts or subscripts can
lead to confusion in the following text, particularly in
interpreting exponents.
Abstract
The Internet offers the possibility of distributing a
molecular modeling task over a number of geographically diverse
computers. If these computations can be arranged not to intrude
on the extant workloads of home and office desktop computers,
vast resources may become available. This scenario offers
extremely limited communication bandwidth, so the problem of
partitioning molecular modeling (normally a fairly
communication-intensive task) in a reasonably efficient manner
becomes interesting. This paper presents a partitioning approach
that will allow individual processors to simulate thousands of
time steps before requiring communication with other processors.
Communication bandwidth is replaced by duplicated
computations, exploiting the spatially localizable influence of
each atom upon its neighbors. Processors are assigned to model
overlapping cubical regions of space. Duplicate modeling results
for overlap regions gradually diverge, due to differences in the
boundary information available to neighboring processors. By
characterizing these divergences and keeping track of them,
processors can update one another locally, each processor acting
as the expert on a small cubical region of space in the center of
its own initially assigned area.
Each processor is ignorant of the world beyond its own
initially assigned region. The unknown effects of that outer
world propogate inward from the known border over time. The
velocity of propogation is characterizable. A processor may
continue computing without communicating with its neighbors until
noticeable external effects reach its area of expertise. With
periodic updates from its neighbors, a processor should be able
to simulate for long periods of time.
The trade-off of communication bandwidth with duplicated
computations is acceptable in the envisioned scenario. This
approach might also be useful in more traditional computing
environments of networked workstations, in cases where
communication bandwidth may be significantly expensive compared
to computation.
Motivation
As work in nanotechnology progresses, interesting designs will
grow to involve millions, perhaps billions, of atoms. There will
be worthwhile task in molecular modeling that would unreasonably
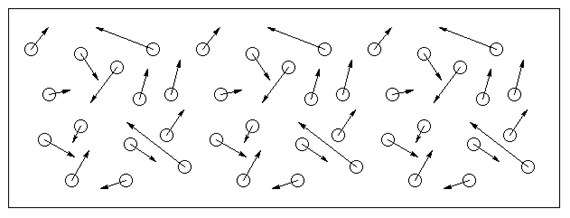
task the resources of a single processor. The illustration below
represents such a task.
Numerous proposals have circulated over the past several
months to perform molecular modeling in a distributed fashion on
the Internet, by borrowing processor cycles from otherwise idle
computers. An obvious approach to parallelizing the computation
involved is to partition the task spatially, and assign a
processor (or some fraction of the attention of a processor) to
each region. This approach is illustrated below.
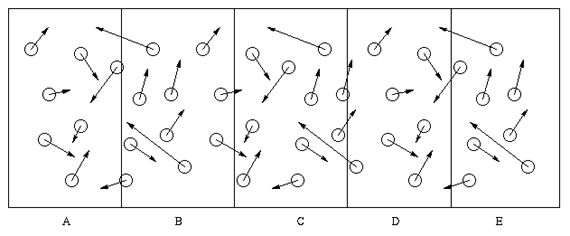
Such proposals are spurred in part by the success of various
other cooperative distributed computations, notably searches of
cryptographic key space. Cryptographic key searches are deceptive
in their success. They are unusual among parallelizable computer
programs in that they require little or no communication between
processors. The difficulty of configuring a problem for parallel
execution is highly dependent on the ratio of communication to
computation performed by each computing node.
It turns out that molecular modeling, partitioned in the most
obvious way illustrated above, would involve a burdensome level
of communication. Because atoms may interact over distances of
several nanometers, pairs of adjacent processors would need to be
in constant communication during the progress of a simulation,
exchanging large quantities of data at every time step. The
situation is aggravated by the fact that atoms may freely wander
from one region to another, requiring processors to renegotiate
ownership of such atoms. Realistic simulations require
sub-femtosecond time steps, and would desirably run for at least
picoseconds and perhaps nanoseconds. The communication overhead
involved would be likely to hopelessly cripple any Internet-based
modeling effort based on the most obvious partitioning; see
Appendix A.
The challenge, then, is:
to design a partitioning that allows terse,
infrequent communication between processors.
Infrequent in this context means at most every few
thousand time steps. In order to design such a partitioning, it
is necessary to examine how algorithms are partitioned for
parallel execution, and also to consider some of the physical and
computational characteristics of molecular modeling.
Partitioning algorithms for parallel computation
In partitioning a problem for parallel execution, it is
generally desirable to choose a partitioning that minimizes
communication between processors. All forms of inter-processor
communication involve non-trivial overhead, including both the
logistical demands of communication (forming data into packets,
implementing communication protocols, and so forth) and
handshaking delays (where one processor must wait until another
is ready to send or receive data). The earlier example of
cryptographic key search is an ideal example of minimal
communication between processors; each key can be tested
separately without reference to any other key, so each processor
is assigned a range of key space to test, which it can work on
for hours or even weeks before its results must be compared with
those of other processors.
One can construct a data dependency graph for an algorithm, a
diagram that displays how each quantity or variable in the
algorithm depends upon others. Such a diagram may be drawn as a
network of vertices connected by edges, where each edge has a
direction, or what graph theorists call a directed graph. One can
then reasonably partition the algorithm for parallel computation
by dividing the vertices of the directed graph into disjoint
subsets, with as few edges as possible connecting any pair of
subsets, since each edge represents an expensive communication
event.
If the directed graph is drawn with inputs primarily on the
left and outputs primarily on the right, then the horizontal
dimension can be thought of as representing time. In this case,
vertical partitions will create vertex subsets that are grouped
temporally, which suggests pipelining as a parallelization
scheme. Horizontal partitions (assuming they are not crossed by
many graph edges) will tend to partition the problem along
spatial lines.
If one draws a dependency graph for a simulation of molecular
mechanics, dependency edges run densely along the time dimension.
A pipelining approach offers little opportunity to minimize
communication. But with the avoidance of electrostatic monopoles,
and the resulting spatial localization of interatomic forces, it
is reasonable to partition a simulation spatially.
Propogation of information within a molecular
modeling task
If I have not seen as far as others, it is
because giants were standing on my shoulders.
-- Hal Abelson
Mathematical models of molecular behavior vary in both
accuracy and computational demand. At the high end, solutions are
sought to Schrodinger's wave equation, indicating positional
probability densities for the electrons belonging to each atom in
the molecule. The most accurate (and computation-intensive) among
such techniques is ab initio modeling, which avoids
entirely the use of any empirically determined physical data,
deriving everything, save for initial positions and velocities,
from first principles. At the low end, simple mechanical models
of molecular behavior employ masses and non-linear springs
[Allinger 1977, Drexler 1992]. Mathematically, the model consists
of a series of terms, each specifying potential energy as a
function of some geometric property of the molecule (an
interatomic distance, an angle between two bonds, or a dihedral
angle involving three bonds). The sum of potential energies is
taken as the potential energy for the entire structure (i.e. the
energy required to deform it from its minimum-energy
configuration to the present configuration), and the negative
gradients of this energy with respect to the positions of the
atomic nuclei are the forces assumed to be acting on those
nuclei.
The principles that motivate the pyramidal partitioning scheme
derive from the physics of atomic interactions as they are
currently understood, and do not depend on whether the modeling
algorithm involves solutions to the wave equation or simple
mass-spring approximations.
For the simulations in this paper (used to obtain
approximations of the speed of sound in cumulene rods), the
mass-spring modeling approach has been employed, since only
approximate results are required. The potential energy formulas
used in simulations for this paper are given in Appendix B, and
are typical for modeling molecular mechanics.
How fast can information move within a molecular dynamics
simulation? Information in this context refers to
information that would influence the positions and velocities of
atomic nuclei. Analogously, one might ask how quickly errors can
propogate through a simulation.
Einstein's theory of relativity states that information cannot
travel faster than the speed of light without incurring
violations of causality. An event in spacetime can only be
affected by other events lying within a four-dimensional cone
aligned along the time axis, which has a spherical
three-dimensional cross-section in space. As time proceeds
backward, this sphere grows linearly at the speed of light.
Similarly, the future events which can be affected by the event
must all lie within a future cone, analogous to a sphere that
grows outward at the speed of light from the event's location in
spacetime. The diagram below illustrates light cones using
two-dimensional space (space being represented by the horizontal
plane and time being represented as the downward-pointing
vertical axis). In the terminology of relativity, these are
referred to as light cones.
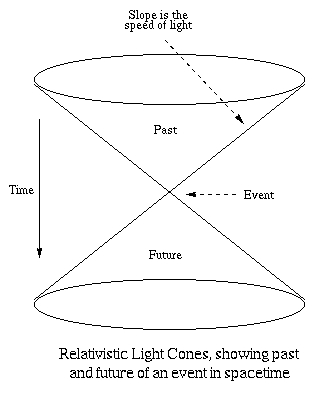
The speed of light places a physical limit on the velocity at
which information may propogate through a molecular dynamics
simulation. In order to compute the position and velocity of all
the atoms in a region from other information available in the
past, we need only consider past events lying within the past
light cone for the region of interest. In practice, information
propogates much more slowly than the speed of light, but it is
heart-warming to have a guaranteed fundamental upper bound.
A more practical upper bound is set by the mechanisms whereby
atoms influence one anothers' trajectories through spacetime.
Physicists and chemists have enumerated a number of forces that
one atom can exert upon another. Covalent bonds involve very
localized forces operating over at most a fraction of a
nanometer. London dispersion forces can act over slightly longer
distances. Electrostatic forces can act over greater distances
yet. All these forces decrease as distances grow large.
For the practical purposes of molecular modeling, it is
customary to neglect sufficiently small forces. This is done by
setting a distance, called the cut-off radius, at which it
is assumed that all direct interactions between one atom and
another would involve negligible forces.
There are also two forms of indirect interaction. An atom may
influence another distant atom at a future time by causing a
chain of influences, each acting locally, but propogating outward
from the first atom to eventually influence the trajectory of the
second. That is, mechanical disturbances may propogate through
the structure. This is simply sound, and the velocity at which
such influences propogate is the speed of sound in the material.
An atom may also influence another atom at a future time, even
though it is more than one cut-off radius away from it, by having
a velocity that carries it toward that atom. In a
nanotechnological design, it can be presumed that significant
velocities acting over significant distances (as opposed to high
velocities that might occur in the normal thermal vibrations of
atoms) have been consciously designed. It is probably a prudent
design practice to require that the intended velocities of moving
mechanical parts in a design should not exceed the speed of sound
in the material. For the purposes of the remainder of this paper,
it will be assumed that this practice has been followed, and that
the speed of sound therefore dictates the maximum velocity at
which mechanical influences can propogate over long distances.
Together, the cut-off distance and the speed of sound offer a
predictable upper bound on the rate at which errors may propogate
through a modeling simulation. Taking this reduced upper bound as
the slope of the cone walls (rather than the speed of light, as
in the relativistic case), one can draw a new diagram
illustrating how the future (the final conditions of the
computation) is affected by the past (the initial conditions of
the computation), yielding a far more feasible computation. In
the actual computation, the sphere-based four-dimensional cone is
replaced by a cube-based four-dimensional pyramid, as cubes can
be packed efficiently.
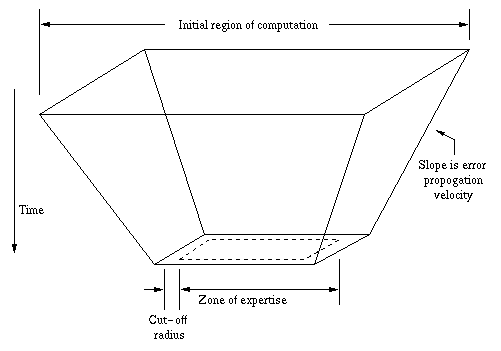
The present proposal assumes that each processor begins with
the initial conditions for a relatively large region of space,
but at the end of a period of simulation time, is responsible for
accurate conditions in a smaller area of expertise. The
purpose of beginning with the larger set of initial conditions is
so that the processor will have all the information required to
arrive at the final conditions in the area of expertise without
consulting other processors. The course of such a computation
over a period of simulation time is illustrated above. The
inverted pyramid plays a central role in the partitioning scheme
presented herein, which is therefore named pyramidal
partitioning.
Earlier, the possibility was mentioned that processors might
need to renegotiate ownership of atoms that wander from one area
of expertise into another over the course of a simulation run. In
the present scheme, this is unnecessary. Each processor has all
the information it needs for the entire time period being
simulated, and it will determine which atoms it owns at the end
of the simulation period.
Optimal pyramid size
Optimal pyramid size is a function of the cost ratio of
communication to computation. At the practical limit, where
computation is essentially free, the initial cube (the top
"surface" of the 4D pyramid) has a linear dimension
three times that of the area of expertise. Mutual updates are
then localized within 3x3x3 neighborhoods.
The two variables that are easily configured are the linear
dimension of the area of expertise and the duration of each
simulation run. The cut-off radius rco
and the error propogation velocity ve
are set by the structure being modeled and, to some extent, the
modeling approach. A very crude measure of the computational cost
of modeling is the integral of the simulated volume over the
simulation time. If the linear dimension of the area of expertise
is S and the duration of the simulation is T, then
this measure can be obtained by evaluating the definite integral.
In MKS units, its units are m3 sec.
C = (1 / 8 ve) [(S
+ 2 rco + 2 ve
T)4 - (S + 2 rco)4]
If computations were restricted only to the area of expertise,
the expression above would reduce to S3
T. The degree to which it exceeds this quantity is a measure
of the duplication of computations throughout the entire network
of screensavers.
In order for this measure to accurately represent
computational cost, the simulated object would need to have
uniform density and structure throughout the simulated region.
This will likely never be the case for an interesting design.
Nevertheless, this measure is likely to be a useful heuristic. It
would need to be multiplied by an empirically determined constant
of proportionality, which would have MKS units of I m-3
sec-1, where I represents a unit of
computation such as a computer instruction or a processor clock
cycle.
For the screensaver case, where MIPs are essentially free and
communication is costly, it makes sense to set S = rco
+ ve T, which gives 3x3x3 cube
packing. Approximately 90% of the computations performed by any
screensaver are duplicated, or nearly duplicated, by others, due
to the overlap of initial cubical regions. Because different
screensavers have access to different information, duplications
will not be exact, but the divergent regions will fall outside
the areas of expertise.
In general, a more detailed cost analysis will offer a better
use of resources. A useful parameter for such analyses is (S +
2 rco + 2 ve T)
/ (S + 2 rco), the ratio of the
initial cube size to the final cube size. For convenience, call
this the pyramid ratio. The initial overlap between cubes
will determine how much information must be exchanged at each
mutual update step. Smaller pyramid ratios correspond to smaller
overlaps, reducing the size of messages but increasing their
frequency. As computation grows more expensive and communication
becomes cheaper, lower overall costs are achieved with lower
pyramid ratios because fewer computations are duplicated.
An important consideration in choosing an optimal pyramid size
is the available hard disk space for the individual machines.
Disk space places an upper bound on the number of atoms a machine
can handle at the beginning of a run. To compute disk space
requirements, assume that each atom requires position and
velocity vectors, as well as a few bytes representing element,
charge, and hybridization. These latter may be representable as
bit fields within a single byte. Assuming vector components
require 8 bytes each, this makes about 50 bytes per atom. With
reasonably good compression, it may be possible to get down to 10
bytes per atom. Assuming that the average computer user is
willing to dedicate 107 bytes of disk space to the
simulation, each computer can model at most 106 atoms
at a time. In a fairly dense material, the spatial density of
atoms would be on the order of 1029 atoms per cubic
meter. At this density, 106 atoms occupy a cube with a
linear dimension of roughly 21 nm. This is the size of the
initial computing region, (S + 2 rco
+ 2 ve T). Assuming 3x3x3 packing,
it will be desirable to assign a value of 7 nm for S.
In the absence of ionized atoms (acting as electric
monopoles), an acceptable cut-off radius is 5.4 nm (see appendix
C). Setting S + 2 ve T + 2 rco
= 21 nm, with rco = 5.4 nm and S
= 7 nm gives ve T = 1.6 nm. In
appendix E, an upper bound on ve of 4x104
m/sec is derived, giving T = 4x10-14 sec. Forty
femtoseconds' worth of simulation is a very reasonable amount of
work to ask individual processors to do in the time between
communications.
This figure assumes 3x3x3 packing. If the rate of
communication is unreasonably slow (e.g. a flurry of emails every
six weeks) then the situation could be improved by reducing the
pyramid ratio. Still assuming an iniitial cube size of 21 nm, it
may be preferable to run a shorter simulation time, perhaps 2.5
fs, between updates, giving a linear dimension for the final area
of expertise of 10 nm rather than 7 nm. Using the measure of
computational cost defined above, the cost of the 40 fs
computation is 2.94x10-37 m3 sec, while the
cost of the 2.5 fs computation is 1.43x10-38m3
sec. The 16-fold reduction in simulated time has yielded a
20-fold reduction in workload. Over a large range of simulated
times, this relationship exhibits this near-linearity.
As communication becomes less expensive, smaller T
values offer the advantage of less duplication of computations.
The cost of duplication can be computed by assuming that an
entirely non-duplicative computation would operate for the full
simulation time over only the volume (S + 2 rco)3,
which works out to 2.3x10-37 m3 sec in the
40 fs simulation, and 1.43x10-38 m3 sec in
the 2.5 fs simulation. Dividing costs by these numbers gives
measures of the cost associated with redundant computation, 1.28
in the 40 fs case and 1.014 in the 2.5 fs case. Apparently the 40
fs case is twenty times as "wasteful" as the 2.5 fs
case.
The optimal trade-off depends on the latency of communication
and the difference in execution times for the different
processors (which in turn depends on processor speed as well as
density variations in the simulated material). It is probably
worthwhile to do more complete cost analyses on a case-by-case
basis.
Recursive pyramidal partitioning
It is useful to be able to subpartition each processor's task
into individually computable subtasks, for two reasons. Assuming
this partitioning scheme is used in a screensaver context, it
must be possible to interrupt the computation at any point and
return the use of the machine to the user. This must be done in
no more than a few seconds at most, and the loss of work in such
a case should be acceptably small. This suggests subtasks that
can be completed in five or ten minutes. The second reason for
subpartitioning is that, while the set of initial conditions for
a machine's entire task might fit on the hard disk, it may not
fit into the machine's RAM. It would then be impossible to
perform the entire task as one job. Even if the entire task fit
in the machine's RAM, it might inconvenience other tasks running
on the machine, or cause more swapping to disk than is
preferable.
The notion of pyramidal partitioning once again comes to the
rescue. The large 4D pyramid can be broken into smaller
overlapping 4D pyramids, whose lower surfaces again form a
cubical lattice, and whose upper surfaces overlap as required to
minimize dependencies between subtasks. Again, the slope of the
sides of the small pyramids is the speed of sound in the
material. The large pyramid is partitioned along both spatial and
temporal dimensions, providing subtasks that do not unreasonably
tax the machine's available memory, nor do they each run more
than about ten minutes. The precise dimensions of the smaller
pyramids would be determined using a similar sort of cost
analysis as was applied to the large pyramids, but with different
constraints reflecting the different role of the small pyramids.
Implementation
At the beginning of a simulation, all machines are given
initial conditions for their initial regions of computation (a
cube with a linear dimension of S + 2 rco
+ 2 ve T in the general case, or 3
S in the 3x3x3 packing case). This information should include
the list of atoms, with initial positions and velocities for each
atom, as well as element and orbital hybridization. Once the
machines have all computed for a time T, they engage in an update
cycle where each uses the updated information from its own
area of expertise to provide new initial conditions for its neighbors,
the 26 machines with adjacent areas of expertise. In the general
case, these updates will describe slab-shaped regions for the six
side neighbors, beam-shaped regions for the twelve edge
neighbors, and small cubes for the eight corner neighbors. In the
case of 3x3x3 packing, these shapes each grow to the entire
cubical area of expertise for that particular neighbor.
Screen saver programs have components that are highly
architecture-dependent. They generally perform graphics
operations at a low level, and in this case, there is a premium
on efficient graphics operations to leave processor time
available for modeling computations. They also need some kind of
timeout on the keyboard and mouse to determine that the machine
is not in use, and a graceful way to abort partially completed
tasks when the machine is needed again. The primary development
should be targeted at the most popular platform, which is
probably an Intel-based computer running Microsoft Windows 95.
The screensaver development effort should draw upon expertise in
platform-specific programming.
Before the public deployment of the screen saver, the
partitioning and communication algorithms can be simulated on a
single machine. This is an opportunity to use debug statements
and monitor simulated email traffic. The program should be
thoroughly debugged before prior to deployment, as subsequent
upgrades will pose significant logistical difficulties. It is
particularly important to resolve any gridlock issues in
simulation (situations where two or more processors are stuck,
each waiting for a message from one of the others). It should be
possible to resolve these issues if processors do not wait for
incoming email before transmitting their own updates and
notifications.
In a system employing geographically diverse machines owned
and operated by different people and organizations, redundancy
must play an important role if a large-scale computation is to
extend over a lengthy period of time. Every area of expertise
should therefore be covered by at least two desktop machines,
which have email addresses for each other. Siblings are
machines that are working on the same area of expertise. The use
of siblings offers both fault tolerance and increased speed. When
a machine finishes a computation, it notifies its siblings that
it has finished and it is transmitting updates to its neighbors
and the central controller. If a machine receives a sibling
notification before it finishes, it should notify its siblings
that it is aborting its own computation, but is still alive. On
each update cycle, each machine should expect to receive from all
its siblings either one of these two types of notifications. If a
notification hasn't arrived by the following update cycle, the
machine should assume that its would-be sender is dead, and email
the central controller with this news. It should cut all ties to
the presumed-dead machine. The central controller may reinstate
the sibling relationship at a later date, or it may assign a new
sibling. The central controller has the luxury of being able to
handle complex situations on a case-by-case basis (e.g. an
operator may telephone the owner of the presumed-dead machine and
confirm its status) but the screensaver itself must only attempt
to handle situations that can be addressed with its own limited,
automated resources.
An unprotected network of busily computing screensavers will
offer a tempting target to hackers. An easy attack would be
simply to spoof system emails, impersonating siblings, neighbors,
and the central controller. Such attacks can be prevented by the
use of digital signatures. The central controller should use
public-key cryptography to sign its outgoing emails. Each
screensaver has a copy of the central controller's public key,
which can be used to authenticate controller emails, but cannot
be used to deduce the controller's private key. Each
sibling-sibling or neighbor-neighbor pair should engage in a
Diffie-Hellman key exchange when the pair relationship is first
established. The effect of doing so is to produce a secret key
known only to the paired machines. This key can be used for
digital signatures of all email between the pair. New
Diffie-Hellman keys are established whenever pairs rearrange, for
example when a machine goes down and must be replaced.
Alternatively, each machine may generate a public-private key
pair when the screensaver first runs, in which case public-key
digital signatures can be used throughout the system.
Software that implements this partitioning scheme will contain
components that would be useful for other tasks. It would be
prudent software engineering to modularize the design with an eye
to reuse. Communication resources can be written as a library, as
can digital signature resources, which could be linked to other
computation-intensive tasks as required. Minimally, it should be
easy to upgrade the molecular modeling algorithms. Force fields
are often designed to specialize in different branches of
chemistry, some better suited for proteins, others for crystals,
and so forth.
A screensaver might not have its computational task
predetermined at compile time, but instead could download it via
email or network. The screensaver could recognize and trust any
of a set of central controllers for whom the computer's owner had
collected public keys, which the screensaver would use to verify
digital signatures. The screensaver might monitor use of
computing and storage resources and present the central
controller with a monthly bill. One can imagine a market economy
in which screensavers accept a set of preferences from their
computers' owners and allocate idle computing time according to
those preferences, while many "central controllers"
attempt to outbid one another for computing resources. In such an
economy, one could collect a small income merely by virtue of
owning a computer (or a large income by owning many computers).
There would be some important security issues to resolve, similar
to those faced by the designers of the Java language.
Other tools
This paper has described a partitioning method that makes it
practical to distribute a molecular modeling task over a wide
number of geographically diverse desktop computers. Other
programs have been proposed to serve this purpose in the context
of a conventional network of Unix workstations, notably the work
of Brunner et. al. (1994-97). The present proposal is likely to
run into difficulties when confronted with electric monopoles, as
the cut-off radius would grow large, necessitating pyramidal
partitions that might be too large to practically fit on a
workstation's hard disk (unless the workstations were dedicated
strictly to this task, or the structure involved were primarily
empty space). Brunner et al. employ the Distributed Parallel
Multipole Tree Algorithm due to Rankin (1997), which claims to
compute electrostatic interactions in linear time. This algorithm
is worthy of further investigation.
Another noteworthy effort is the Condor program (Basney et.
al., 1997). Like the proposed screensaver program, it uses a
machine's idle CPU cycles for large computational tasks, and
coordinates groups of machines. Having learned of this program
shortly before this paper was due, the present author has not
been able to research Condor as thoroughly as it deserves.
To make such a system useful overall, there would need to be
other components that have not been addressed here. Two of these
are a means for generating structures and a means for visualizing
the results of simulation runs to verify that the intended
behavior occurred. Structure generation is a potentially rich
area of investigation; interesting structures can be fully as
complex as large computer programs, and many methods for managing
complexity developed in computer science may find applicability
in rational structure design. Krummenacker (1993) has explored
the possibility of structure generation in Common Lisp,
exploiting that language's ability to manage both complexity and
abstraction.
Visualization programs abound; some examples are RasMol,
pdb2pov, moviemol, and VMD. It is also possible to combine the
functions of structure generation and visualization into a single
program (as in the present author's own effort NanoCAD, and the
commercial program Molecules 3D) although this approach is
unlikely to scale well to structures of more than a few thousand
atoms, and it offers no opportunity to manage complexity in the
computer science sense. It is useful to divide the
functionalities of structure generation and visualization,
connecting them by a common file format; this allows for
independent innovation in both areas.
Catalogs of standardized components simplify the design work
of present-day electronic and mechanical engineers. Another area
that this paper has not touch upon is the compilation of
structure libraries. A few libraries exist on the web already,
such as the Brookhaven Protein Databank, but none are yet
particularly suitable as nanotechnological parts catalogs. A
parts catalog should include information for each entry such as
physical size, gross physical characteristics (perhaps a crude
computational model of its mechanical characteristics, along with
information about charge distribution) and some form of
visualization, as well as recommendations by the part's designer
regarding intended uses, caveats, references to previous designs,
and so forth. It is possible that eventually such parts would be
reviewed, as many products are today reviewed in magazines. This
would facilitate the function of a free market for
nanotechnological component designs.
Appendix A: Assumed computing environment
The computing environment envisioned for this paper assumes
that portions of the modeling task are performed by screensavers
which communicate via email every few days. This is in contrast
to some scenarios involving the distribution of computational
load via Java applets.
The Java applet scheme would seem to suffer from some serious
drawbacks. First, the web page from which the applet is
distributed must draw substantial interest on the web over an
extended period of time. It is difficult to imagine a web page
with content so compelling that it would attract the hundreds or
thousands of visitors necessary to make the effort worthwhile.
Second, each visitor is likely to visit the web page for only a
short period of time, so little computation can be expected for
each visit. Third, each instantiation of the applet would need to
communicate with the central host, to receive initial values and
to return computed results. If there are enough applets running
on the web to justify the applet approach, it seems likely that
the central host's ability to communicate, to assign new tasks,
and to accept and process responses would become a significant
bottleneck.
Because a screen saver resides permanently on a computer, it
can maintain state information on disk files and perform
computations over an extended period. The limitation on a screen
saver is its capacity to communicate with similar programs
running elsewhere. Email can presumably be queued, and sent and
received using email software already present on the computer,
along with the user's own email.
It is assumed that the computational resources made available
by the distribution of screensavers is essentially free. This
proposal is actually rather wasteful of those resources. In the
entire network of screensavers, approximately 90% of the work
will be duplicated, or nearly duplicated. This duplication has
been designed to minimize the need for frequent communication.
It is likely that the majority of available desktop machines
would be Intel-based computers running Microsoft Windows 95. When
the time came to write an actual screensaver, that would probably
be the most populous platform and therefore the highest priority
for implementation.
The scheme envisioned in this paper does not require
substantial participation from a central host, once the initial
conditions are set for a lengthy simulation run. Processors
update one another by email a few times each week. These updates
are also sent to the central host, or indeed to anyone interested
in the simulation results.
Appendix B: Modeling molecular mechanics
The approach used here for molecular modeling simulations is
similar to the MM2 modeling method described by Drexler (1992,
page 44). More accurate modeling methods exist, involving the
derivation of solutions to Schrodinger's wave equation to
determine the position probability functions for the electrons in
a structure, but these are much more computationally costly. The
MM2 method gives reasonable approximations for the mechanical
behavior of molecules, by assuming that the forces between atoms
can be modeled as a series of non-linear springs. MM2 and similar
methods have been in use by computational chemists for many
years.
MM2 assigns potential energy functions to various geometric
properties of a group of atoms. Generally, these potential energy
functions are quadratic in the parameter in question, thereby
giving spring-like behavior. Of particular interest for the
simulations in this paper are the energy functions associated
with covalent bond lengths, electrostatic interactions, and van
der Waals forces. Van der Waals forces are generally considered,
for the purposes of computational chemistry, to encompass both
the attractive London dispersion force and steric repulsion due
to overlap of electron clouds. The van der Waals interaction
differs markedly in algebraic form from the other terms used in
MM2.
For the energy associated with perturbations in bond length,
Drexler gives an expression including a quadratic and cubic term,
but notes that this expression is valid only close to the
minimum-energy bond length. He later gives expressions for
over-long bonds based on the Morse function, basically involving
an exponential drop-off.
To handle bond lengths over large ranges, I have chosen an
energy function which follows the quadratic-cubic form close to
the bond's nominal length, and switches to a decreasing
exponential some distance away. I juggled a few coefficients to
get the energy to look reasonably smooth. While this may not be
entirely realistic (as would be the case were I solving the
Schrodinger wave equation), it is adequate for the simulations in
this paper.
Assuming a covalent bond stiffness of ks
and a nominal bond length of r0, I have
used a bond stretching potential energy given by:
E = 0.5 ks 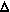 r2
(1 - kc r) - z ks if r < rt r2
(1 - kc r) - z ks if r < rt
E = -(a ks / b) exp (-b r) otherwise
Taking the first derivative yields bond stretching force,
acting in a direction to move the atoms closer to a distance of r0:
F = ks r (1 - 1.5 kc
r) if r < rt
F = a ks exp (-b r) otherwise
where
r
= r - r0
kc = 2.0x1010
m-1
rt = 1.5x10-10
m
b = 3.0x1010 m-1
a = rt (1 - 1.5 kc
rt) exp (b rt)
z = (a / b) exp(-b rt)
+ 0.5 rt2 (1 - kc
rt)
a and z depend on other values and ensure that
the transition from the quadratic-cubic expression to the
exponential expression will be smooth. The value for kc
is supplied by Drexler (1992, page 44). b, the rate of
exponential drop-off, and rt, the radius
at which the switch is made from quadratic-cubic to exponential,
were chosen to give a smooth energy and force graphs over a range
of typical values for bond length and stiffness; this was in
large measure an esthetic judgement and should not be assumed to
have a significant correlation with physical reality.
Here is a plot of the energy function, using the length and
stiffness for the bond between two sp-hybridized carbon
atoms. The horizontal axis is marked in nanometers (10-9
meters) and the vertical axis is marked in attojoules (10-18
joules).
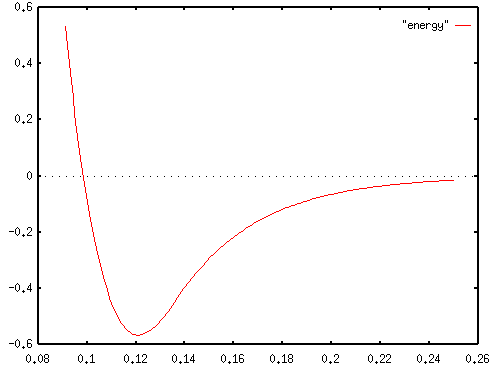
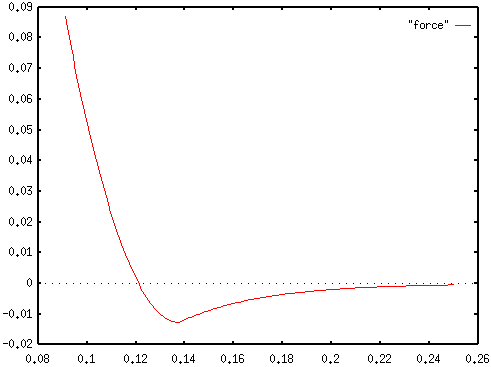
Force is the negative gradient of potential energy. Here,
positive values represent repulsion and negative values represent
attraction. The force is plotted here for the same C-sp-C-sp
bond. The horizontal axis is marked in nanometers (10-9
meters) and the vertical axis is marked in micronewtons (10-6
newtons).
MM2 also supplies potential energy formulas for the angle
between two covalent bonds (involving three atoms) and the
dihedral angle described by three covalent bonds (involving four
atoms). The simulations in this paper involve one-dimensional
structures that do not require these angular energy terms, but it
is important to note that like the expression relating bond
length to potential energy, the effects of these terms is highly
localized.
In addition to forces related to covalent bonds, MM2 uses
conventional formulas for electrostatic attraction and repulsion,
relegated here to appendix B. London dispersion forces and steric
repulsion are usually lumped into a single energy expression,
called the Lennard-Jones potential, typically of the general form
r-6 - r-12.
Partitioning the modeling problem becomes simpler if the
effects of forces can be made more local. A simple way to do this
with the electrostatic force is to insist that all atoms be
non-ionized, or electrically neutral. There are still
electrostatic forces to contend with, but they are now
interactions between electric dipoles instead of electric
monopoles, and they are more local because they fall off more
quickly. Structures in which no atoms are ionized are likely to
be more stable anyway, and therefore of interest in
nanotechnology.
In the absence of ionized atoms, a structure cannot be
ionically bonded (as are ice and salt). All bonds must be
covalent. When a covalent bond forms between atoms of differing
electronegativities, the electron cloud is distributed unevenly.
The electrons flock to the atom with higher electronegativity,
exposing the positively charged nucleus of the atom with lower
electronegativity. As a result, covalent bonds may be regarded in
such cases as electric dipoles. Tables of dipole moments for
covalent bonds have been compiled, and are part of the MM3 model
of molecular mechanics.
Appendix C: Determination of cut-off radii
In order to determine the cut-off radius for one of the forces
discussed thusfar, it is important to determine the maximum
tolerable error in the sum of forces acting on any atom in the
simulation. Then it is fairly easy to solve for the minimum
distance over which a force would need to act in order not to
exceed this error value in magnitude. Once cut-off radii have
been determined for all the forces at work in simulations, the
largest can be used as an overall cut-off radius for the entire
simulation.
All the force contributions in a simulation will be subject to
round-off and quantization errors. Frequently, the coefficients
used in specifying force fields are available to only three or
four significant figures. In deciding a maximum tolerable error
force, it is sufficient to choose a value comfortably below the
level of unavoidable round-off errors.
In a spring-mass molecular mechanics model, the most
significant round-off errors will occur in connection with bond
length energy terms, and of these, the greatest forces will be
associated with strong covalent bonds such as the bond between a
pair of sp-hybridized carbons. This bond has a stiffness
of about 1500 N/m. At room temperature, two atoms bonded in this
way will vibrate with an energy of about kT, or about 4x10-21
J. The amplitude of the vibrations will be approximately 2x10-12
m, with forces of approximately 3x10-9 N. Given that
forces of this magnitude will occur due to random thermal
vibration, it would seem that an error force of 10-13
N should be adequately prudent.
Coulomb's law states that the electrostatic force (Feynman,
1963, page 4-2) acting between monopoles is given by this
formula, where Ke is 8.98755x109
N m2 C-2, q1 and q2
are the electric charges of the monopoles in coulombs, and r
is the distance between the monopoles in meters:
f = Ke q1
q2 / r2
and solving for r, to yield:
rco = sqrt (Ke
q2 / fco) =
(3x1011 m C-1)
q
If q is the charge of a single proton or electron, then
rco works out to 50 nm. This is already
unpleasantly large, and there is no reason q should be
limited to a single charge quantum.
It is possible to do much better by imposing a constraint on
nanotechnological designs (at least those simulated using this
method) that they be restricted to non-ionized atoms, thereby
eliminating electric monopoles. Covalent bonds between atoms of
differing electronegativities still have electric dipole moments,
however, and a cut-off radius must be computed for these.
For electric dipoles, things are far more promising (that is,
the cut-off radius is much smaller). The formula for
electrostatic attraction between dipoles is:
f = 6 Ke d1
d2 / r4
where d1 and d2
are the components of the two dipoles in the r direction.
A reasonable upper bound for d is the largest likely
dipole moment of a covalent bond. Covalent bonds are typically no
more than 2 angstroms long, and the electric charge at each end
will not exceed a proton charge, so d can be expected not
to exceed about 4x10-29 C m. Again solve for r:
rco = (6 Ke
d2 / fco) 1/4
= 5.4 nm
The forces associated with covalent bonds act only between
pairs or small groups of atoms. They can be assumed to have a
cut-off radius on the order of at most a nanometer.
The other long-range interaction of interest is the van der
Waals interaction. By convention, this is actually the sum of two
interactions, the attractive London dispersion force, and steric
repulsion due to the overlap of electron clouds. Drexler (1992)
expresses the potential energy for the van der Waals interaction
as:
Vvdw = evdw
[ 2.48x1015 exp (-12.5 r / rvdw)
- 1.924 (r / rvdw)-6
]
For the purpose of computing a cut-off radius, we seek
worst-case values for evdw and rvdw,
that is, as large as might reasonably be expected. Selecting the
largest values from Drexler's table (1992, page 44) gives evdw
as 3.444x10-21 J and rvdw as
4.64x10-10 m. For these values, the energy expression
becomes:
Vvdw = (8.54x10-16
J) exp [(-2.69x1010 m-1)
r] - (6.61x10-77 J m6)
r-6
The first derivative with respect to r yields the force
due to the van der Waals interaction:
Fvdw = (-2.29x10-5
N) exp [(-2.69x1010 m-1)
r] + (3.97x10-76 N m7)
r-7
At an r value of 10-9 m, the exponential
term contributes only 4.76x110-17 N and decreases
rapidly as r increases. At the same r value, the
second term contributes 3.97x10-13 N, and shrinks
slower than the exponential. For large r, the exponential
term may be safely ignored, and again we solve for rco
in terms of fco:
rco = (1.69x10-11
N1/7 m) fco-1/7
= 1.22x10-9 m
The cut-off radius for van der Waals force is comfortably
smaller than the electrostatic dipolar cut-off radius of 5.4
nanometers. At that distance, the van der Waals force has
diminished to 5x10-19 N and may be neglected. Overall,
a cut-off radius of 5.4 nanometers is acceptable.
Appendix D: The speed of sound
Mechanical disturbances can propogate along chains of covalent
bonds within a material, and this is one of the mechanisms by
which errors may propogate within a simulation. The velocity of
propogation for such disturbances is the speed of sound in the
material. Insight into this mode of error propogation be gained
by examining sound propogation in covalently bonded solids. A
simple scenario for such study is the cumulene rod.
Each atom in the rod is acted upon by forces due to its
bonding interactions with the atoms to its immediate left and
immediate right. Let x represent distance along the rod, x represent the
nominal bond length between adjacent atoms, u represent a
small rightward perturbation in the positions of atoms, m
represent the mass of an atom, and K represent the bond
stiffness between adjacent atoms. u can be considered a
function of x and t (time). Considering only the
quadratic term in the expression for bond length energy, the
force acting upon an atom is given by:
m 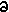 2u/t2 = K [(u(x+x) - u(x)) - (u(x) - u(x-x))] 2u/t2 = K [(u(x+x) - u(x)) - (u(x) - u(x-x))]
The righthand side of this equation resembles the form of a
second spatial derivative, so with reasonable accuracy, the
equation can be rewritten:
2u/t2
= (K x2
/ m) 2u/x2
Equations of this form are called wave equations [Hildebrand,
page 399, equation 64]. Their solutions are of the form:
u(x,t) = f(x - ct) + g(x + ct)
where c = x
sqrt(K/M), the error propogation velocity, and f and g
represent error signals respectively moving rightward and
leftward along the x axis. This is consistent with the
common expression given for the speed of sound in a solid
material, the square root of the radius of Young's modulus
divided by density.
For cumulene,
x
= 1.21x10-10 m
K = 1525 N/m
m = 2.007x10-26 kg
giving an expected propogation velocity of 3.33x104
m/sec. In my simulations, I saw a speed closer to 3.98x104
m/sec. I attribute the difference to the fact that my expression
for the covalent bond energy is not a simple quadratic as assumed
here. The expression I am using is explained in Appendix B. I
believe the simulation result is probably more accurate. Given
the unusually stiff bonds in cumulene, and the fact that they are
all aligned, it is probable that a value of 4x104
m/sec can be used as a reasonable upper bound on the speed of
sound in any structure of interest, and this value has been used
in this capacity elsewhere in this paper.
Another way to express the result derived above would be to
say that each bond in the cumulene rod incurs a propogation delay
of 2.85x10-15 seconds, the bond length divided by the
propogation velocity. If each bond is taken to represent a fixed
propogation delay, then more complex structures such as
diamondoid can be analyzed by summing the propogation delays over
the shortest path of bonds connecting two distant points. It is
worth noting that the resulting velocity need not be anisotropic,
that is, the speed of sound may vary in different directions
through the material.
The speed of sound may also be determined from simulation
results.
In order to model sound propogation, I have chosen to use a
cumulene rod, a covalently-bonded column of sp-hybridized
carbons. The cumulene rod has two desirable properties for such
modeling. Because it is essentially one-dimensional (and because
I am looking for an approximate answer, not an exact one), the
dynamic simulation can itself be done in only one dimension. This
eases the computational burden, simplifies the software involved,
and allows for straightforward presentation of results on
two-dimensional paper. Secondly, it is more densely bonded than
most structures of interest, and therefore provides a worst-case
scenario for error propogation due to chained covalent bonds.
The bond between two sp-hybridized carbons has a stiffness of
1525 newtons per meter and a default distance of 1.21 angstroms.
It is easy therefore to set up a row of sp carbons bonded at
rest, and see what happens when they are struck by another sp
carbon striking them and bonding to the end. The result is
similar to the behavior of the popular "executive toy"
that consists of a row of metal balls suspended by strings: the
momentum is transferred down the row, and finally reaches the
last atom, which separates from the rest and flies off with
approximately the same velocity with which the first atom struck
the row. In the plot below, the horizontal axis is marked in
femtoseconds (10-15 seconds) and the vertical axis is
marked in nanometers (10-9 meters).
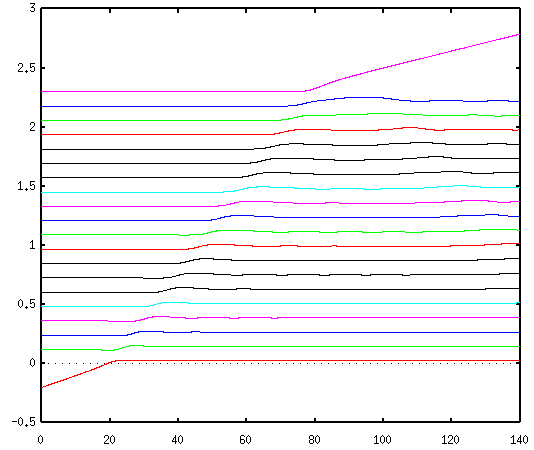
A particularly pretty feature of this plot, typical of
phenomena described by wave equations, is a subtly visible
reflected wave propogating back along the rod.
As expected from the result derived in appendix D, the
propogation velocity is independent of the initial velocity of
the first atom, depending only on the mass of each atom, and the
length and stiffness of the bond.
Appendix E: Application to conventional computer networks
This partitioning scheme arose in the context of
geographically diverse screen savers communicating by email, but
the same fundamental approach could be applied to more
traditional computer networks. The present approach reduces
communication bandwidth at the expense of duplicated
computations. On networks where communication is at a premium but
computation is relatively abundant, the same idea can be applied
with TCP/IP packets instead of email. In fact, the source code
for implementing this approach should be quite similar to the
simulation code used in developing and debugging the screen
saver.
Because this is an approach for scaling a simulation, rather
than a simulation approach per se, it should be applicable with
any currently used modeling software. This is good news for those
who have been using modeling software on one machine, or a few,
but haven't seen how to scale it effectively to much larger
networks. Because the communications discussed here are sparse
and easily characterized, pyramidal partitioning should make it
possible to scale modeling runs almost without bound, with little
new investment in software development.
References
- Basney, J., Cai, W., Greger, M., van der Linden, F.,
Livny, M., Overeinder, B., Pruyne, J., Raman, R., and
Tannenbaum, T. (1997) Condor. http://www.cs.wisc.edu/condor/
- Brookhaven Protein Databank. http://www.rcsb.org
- Brunner, B., Dalke, A., Gursoy, A., Humphrey, B., and
Nelson, M. (1994-97) NAMD. http://www.ks.uiuc.edu/Research/namd/
- Brunner, B., Dalke, A., Gursoy, A., Humphrey, B., and
Nelson, M. (1994-97) VMD. http://www.ks.uiuc.edu/Research/vmd/
- Drexler, K. E. (1988) Rod logic and thermal noise in the
mechanical nanocomputer. Molecular Electronic Devices.
Amsterdam: North-Holland.
- Drexler, K. E. (1992) Nanosystems: Molecular
Machinery, Manufacturing, and Computation. New York:
John Wiley & Sons.
- Elmore, W. C. and M. A. Heald. (1985) Physics of
Waves. New York: Dover.
- Feynman, R. P., R. B. Leighton, and M. Sands. (1963) The
Feynman Lectures on Physics. Reading, Massachusetts:
Addison-Wesley.
- Hildebrand, F. B. (1976) Advanced Calculus for
Applications. Englewood Cliffs, New Jersey:
Prentice-Hall.
- Krummenacker, M. (1993) Cavity Stuffer. http://www.n-a-n-o.com/nano/cavstuf/cavstuf.html
- Ojamae, L. and Hermannson, K. (1997) Moviemol. http://www.chemistry.ohio-state.edu/~lars/moviemol.html
- Molecular Arts Corp. Molecules3D. http://www.molecules.com/
- Rankin, B. (1997) Distributed Parallel Multipole Tree
Algorithm. http://www.ee.duke.edu/Research/SciComp/Docs/Dpmta/dpmta.html
- Sayle, R. Rasmol. http://www.umass.edu/microbio/rasmol/
- Suchanek, E. G. (1995) pdb2pov http://www.intcom.net/~suchanek/pdb2povf/pdb2pov.html
- Ware, W. (1997) NanoCAD, a freeware nanotech design
system in Java http://world.std.com/~wware/ncad.html
|